

Syncmers are more sensitive than minimizers for selecting conserved k‑mers in biological sequences
- PMID: 33604186
- PMCID: PMC7869670
- DOI: 10.7717/peerj.10805
Syncmers are more sensitive than minimizers for selecting conserved k‑mers in biological sequences
Abstract
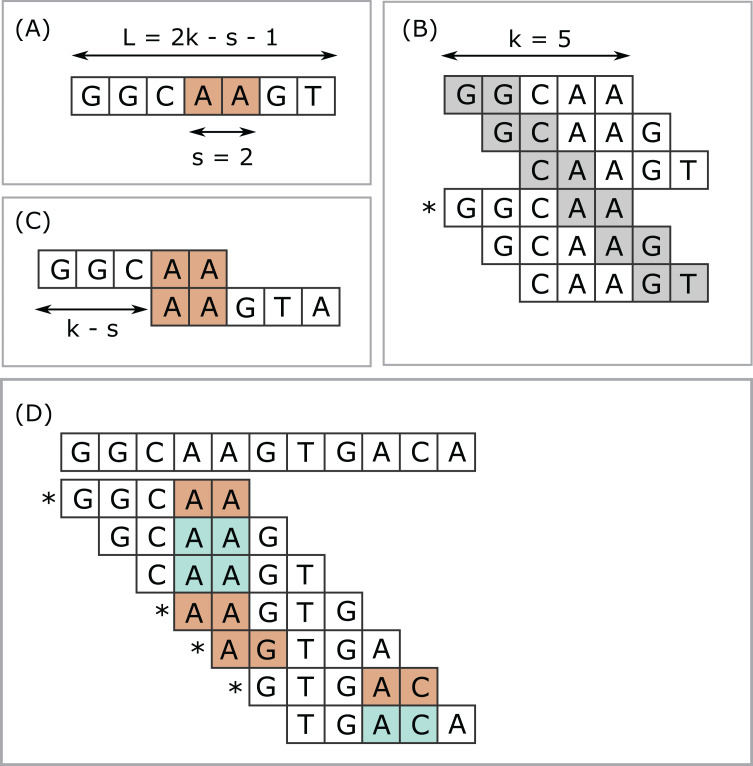
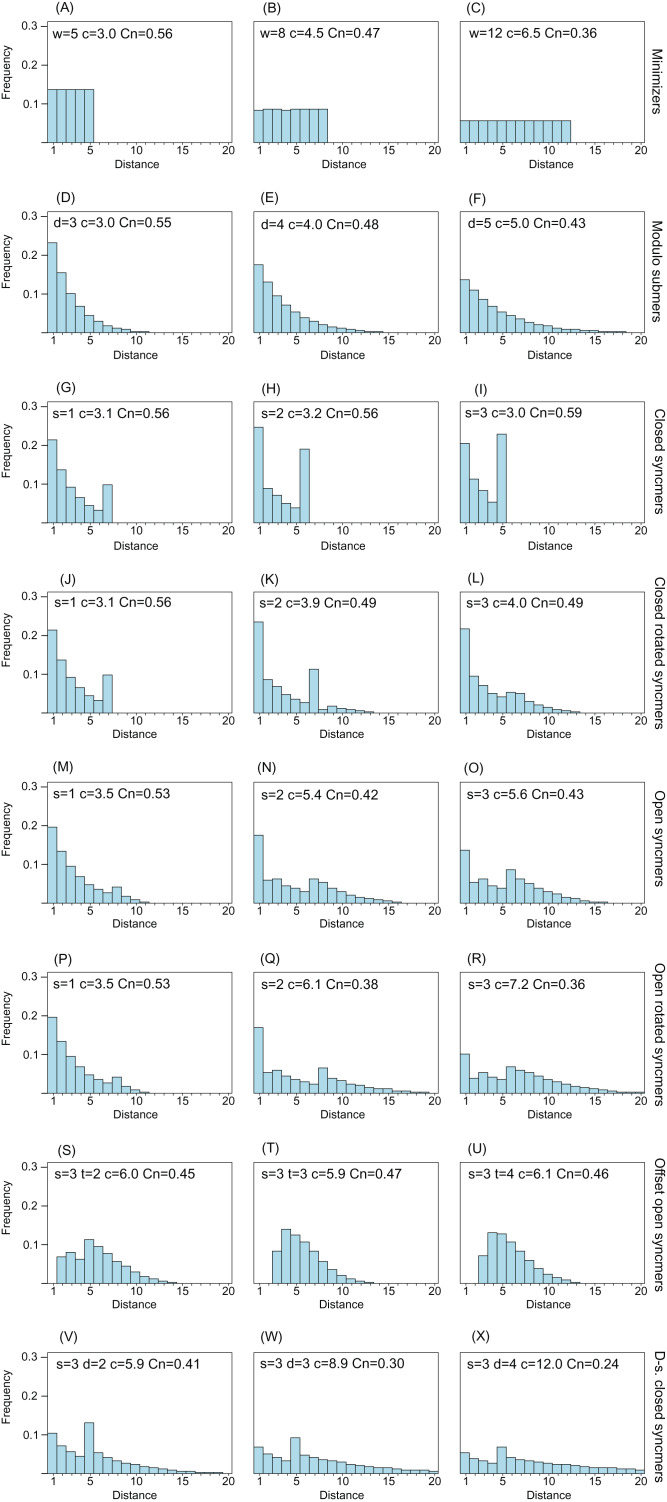
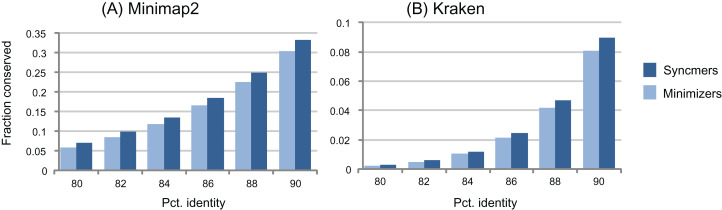
Minimizers are widely used to select subsets of fixed-length substrings (k-mers) from biological sequences in applications ranging from read mapping to taxonomy prediction and indexing of large datasets. The minimizer of a string of w consecutive k-mers is the k-mer with smallest value according to an ordering of all k-mers. Syncmers are defined here as a family of alternative methods which select k-mers by inspecting the position of the smallest-valued substring of length s < k within the k-mer. For example, a closed syncmer is selected if its smallest s-mer is at the start or end of the k-mer. At least one closed syncmer must be found in every window of length (k - s) k-mers. Unlike a minimizer, a syncmer is identified by its sequence alone, and is therefore synchronized in the following sense: if a given k-mer is selected from one sequence, it will also be selected from any other sequence. Also, minimizers can be deleted by mutations in flanking sequence, which cannot happen with syncmers. Experiments on minimizers with parameters used in the minimap2 read mapper and Kraken taxonomy prediction algorithm respectively show that syncmers can simultaneously achieve both lower density and higher conservation compared to minimizers.
Keywords: Alignment-free methods; Minimizers; Sequence analysis; String index; k-mers.
© 2021 Edgar.
Conflict of interest statement
The author declares that he has no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
