

The novel driver gene ASAP2 is a potential druggable target in pancreatic cancer
- PMID: 33605496
- PMCID: PMC8019229
- DOI: 10.1111/cas.14858
The novel driver gene ASAP2 is a potential druggable target in pancreatic cancer
Abstract
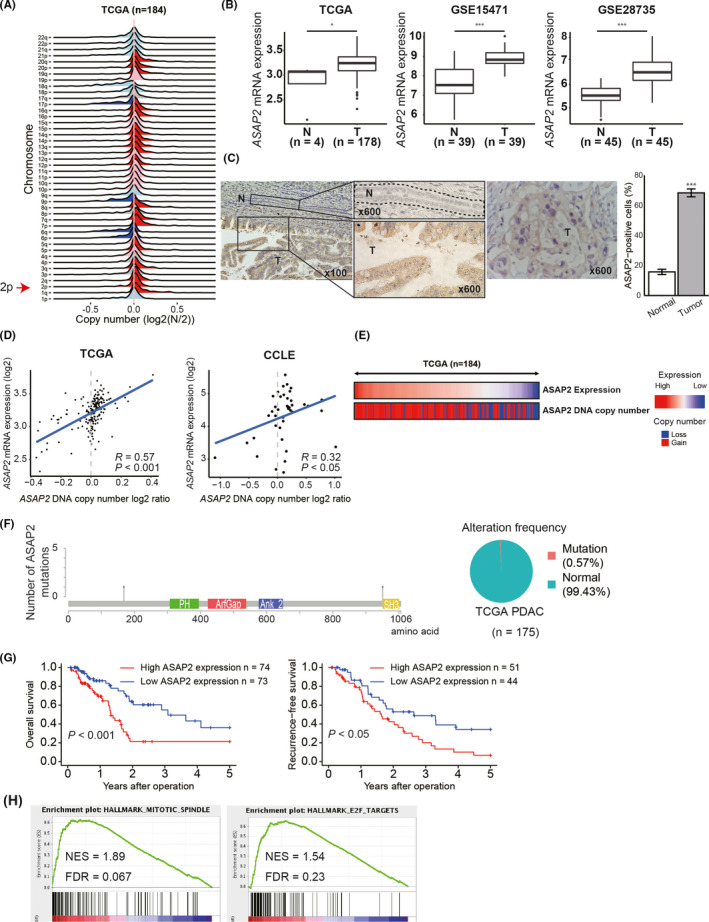
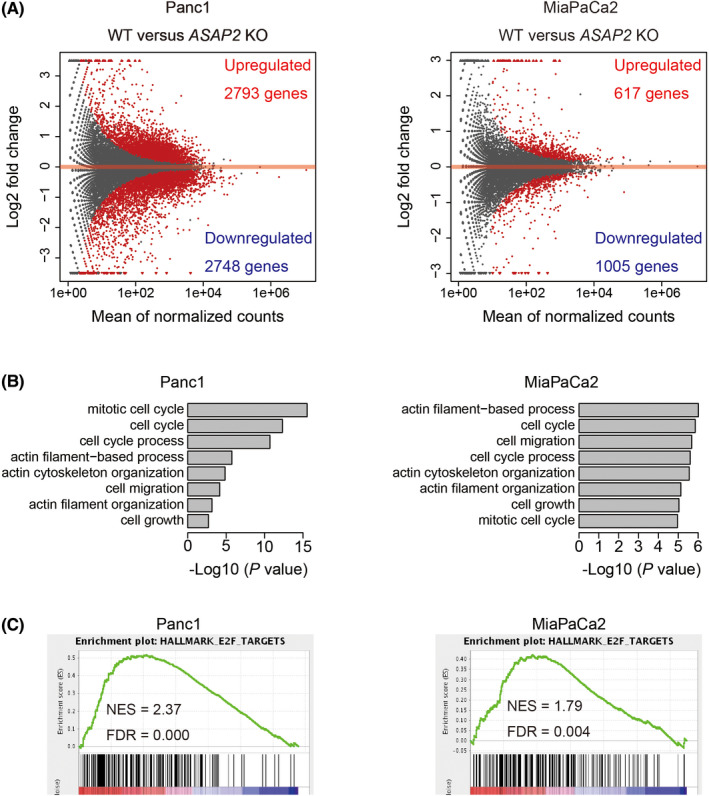
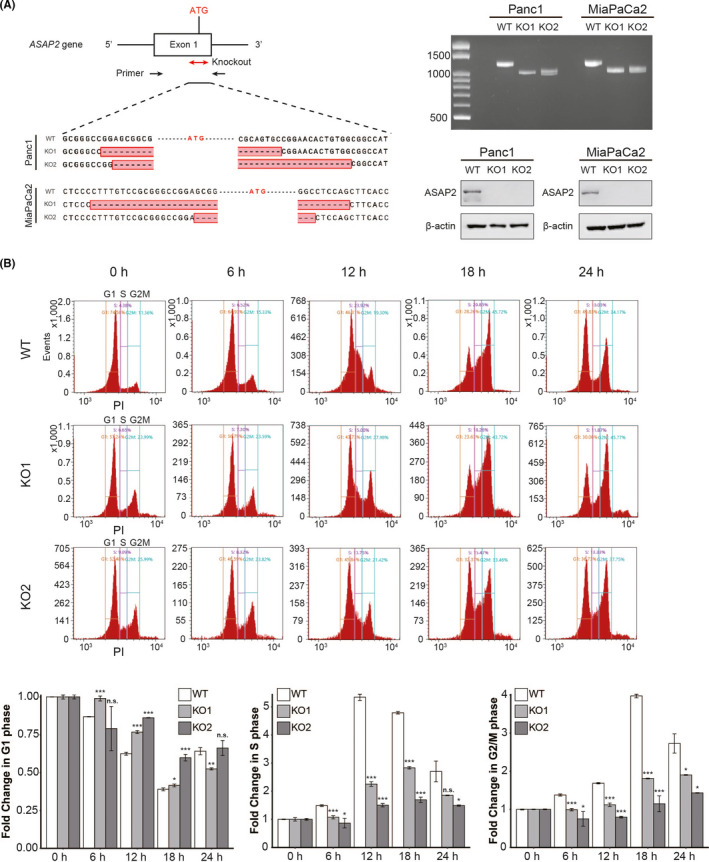
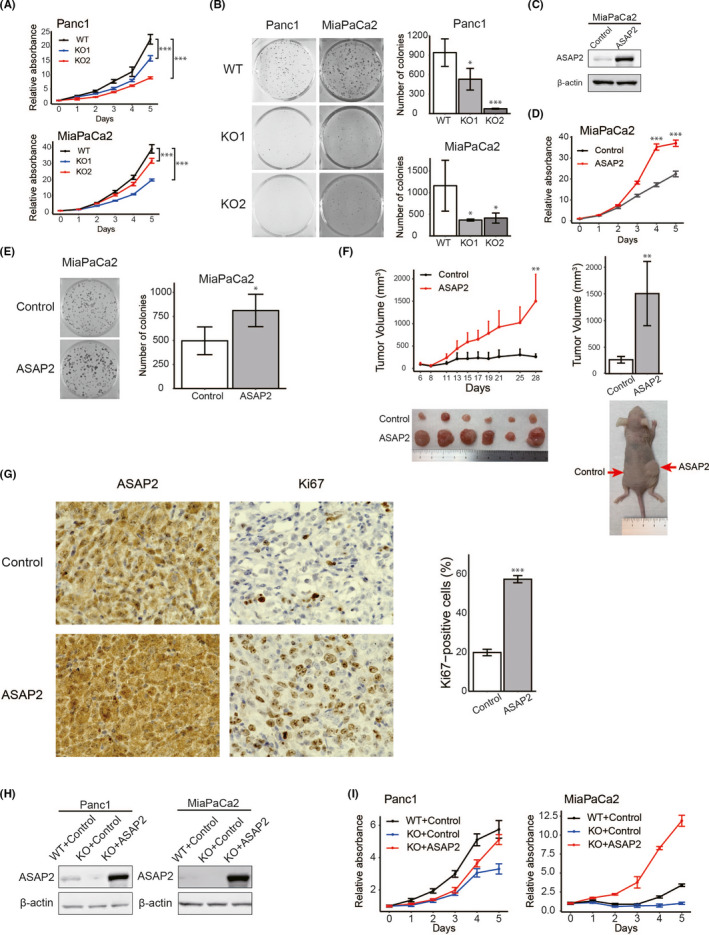
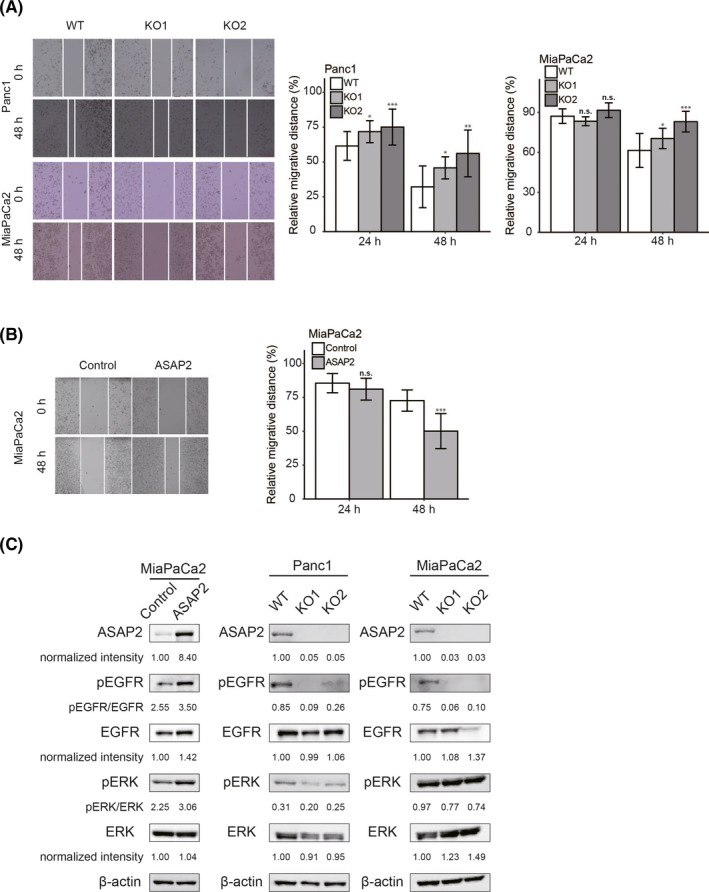
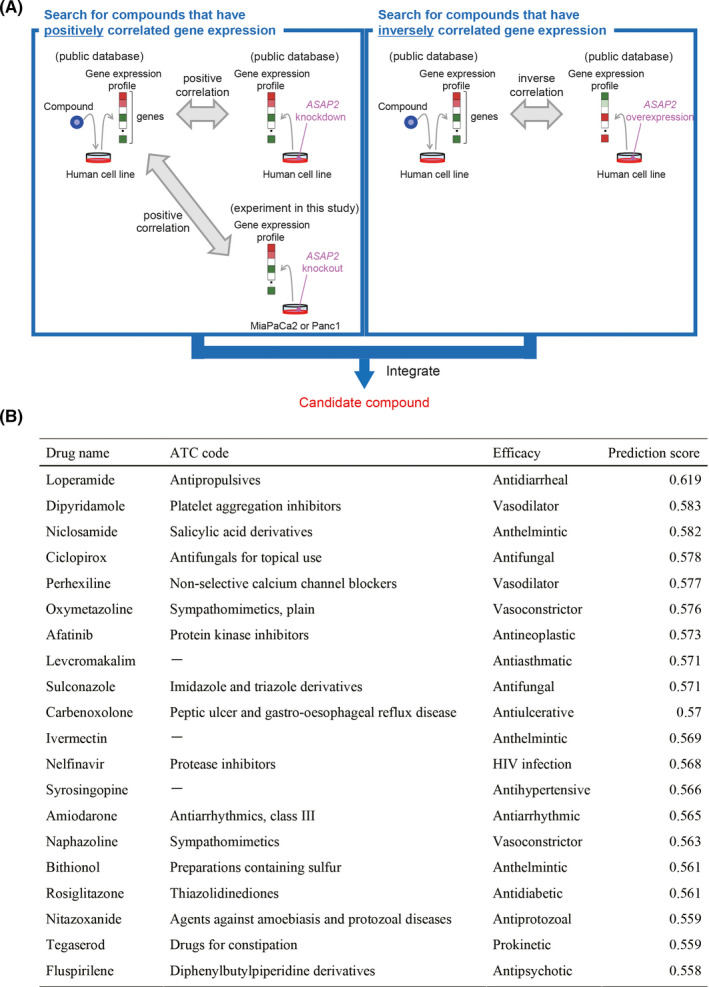
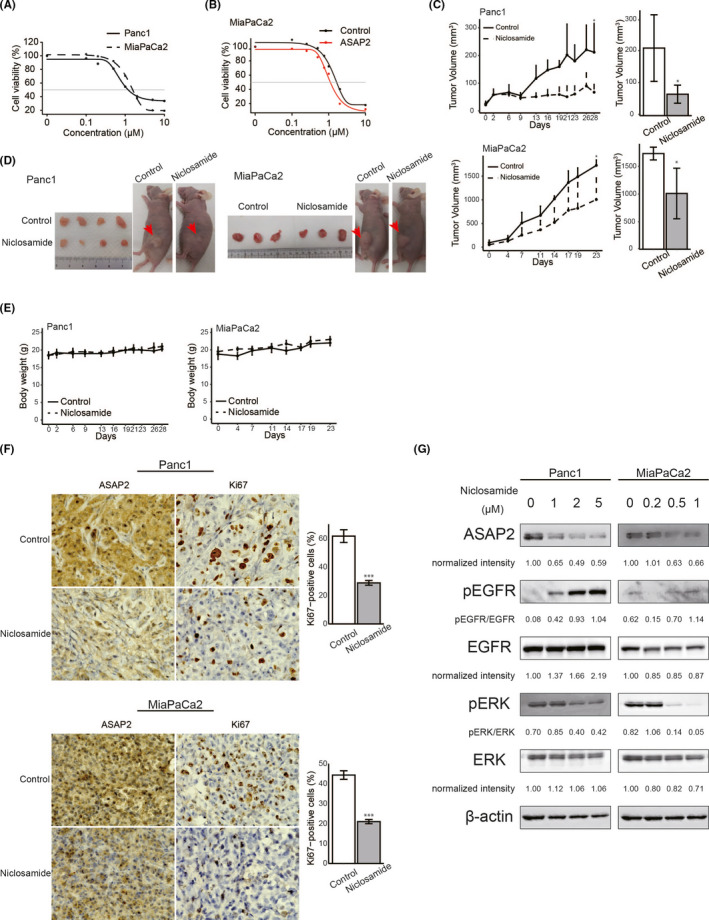
Targeting mutated oncogenes is an effective approach for treating cancer. The 4 main driver genes of pancreatic ductal adenocarcinoma (PDAC) are KRAS, TP53, CDKN2A, and SMAD4, collectively called the "big 4" of PDAC, however they remain challenging therapeutic targets. In this study, ArfGAP with SH3 domain, ankyrin repeat and PH domain 2 (ASAP2), one of the ArfGAP family, was identified as a novel driver gene in PDAC. Clinical analysis with PDAC datasets showed that ASAP2 was overexpressed in PDAC cells based on increased DNA copy numbers, and high ASAP2 expression contributed to a poor prognosis in PDAC. The biological roles of ASAP2 were investigated using ASAP2-knockout PDAC cells generated with CRISPR-Cas9 technology or transfected PDAC cells. In vitro and in vivo analyses showed that ASAP2 promoted tumor growth by facilitating cell cycle progression through phosphorylation of epidermal growth factor receptor (EGFR). A repositioned drug targeting the ASAP2 pathway was identified using a bioinformatics approach. The gene perturbation correlation method showed that niclosamide, an antiparasitic drug, suppressed PDAC growth by inhibition of ASAP2 expression. These data show that ASAP2 is a novel druggable driver gene that activates the EGFR signaling pathway. Furthermore, niclosamide was identified as a repositioned therapeutic agent for PDAC possibly targeting ASAP2.
Keywords: ASAP2; driver gene; drug repositioning; niclosamide; pancreatic cancer.
© 2021 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Conflict of interest statement
The authors declare no conflicts of interest for this article.
Figures
References
-
- Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014;74:2913‐2921. - PubMed
-
- Wilentz RE, Geradts J, Maynard R, et al. Inactivation of the p16 (INK4A) tumor‐suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740‐4744. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
