

LACC1 deficiency links juvenile arthritis with autophagy and metabolism in macrophages
- PMID: 33606008
- PMCID: PMC7901146
- DOI: 10.1084/jem.20201006
LACC1 deficiency links juvenile arthritis with autophagy and metabolism in macrophages
Abstract
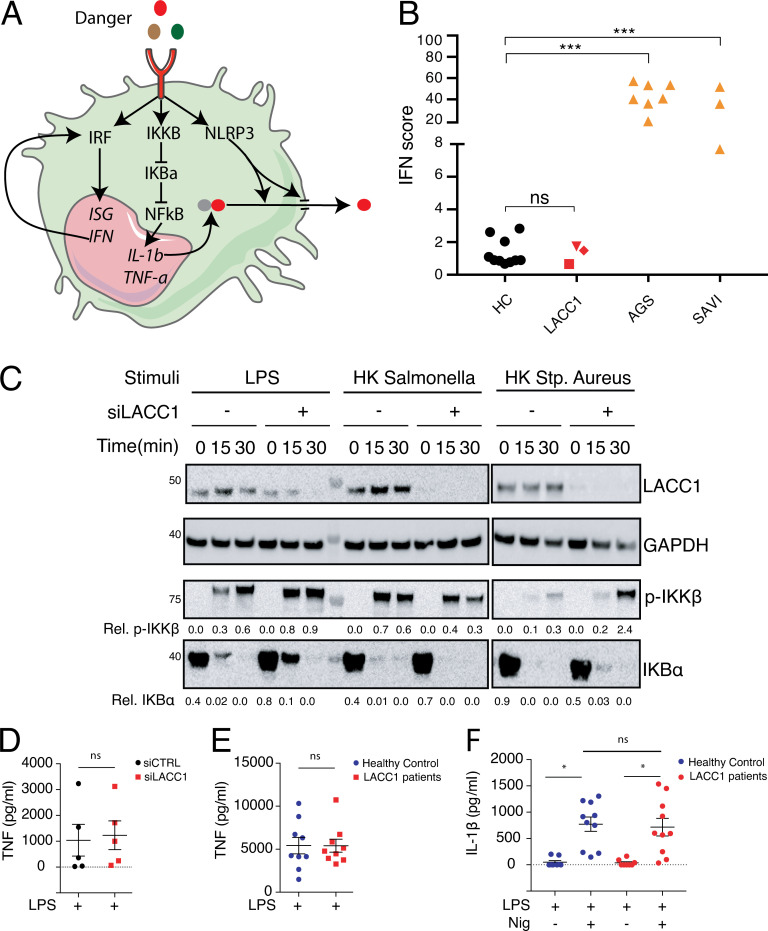
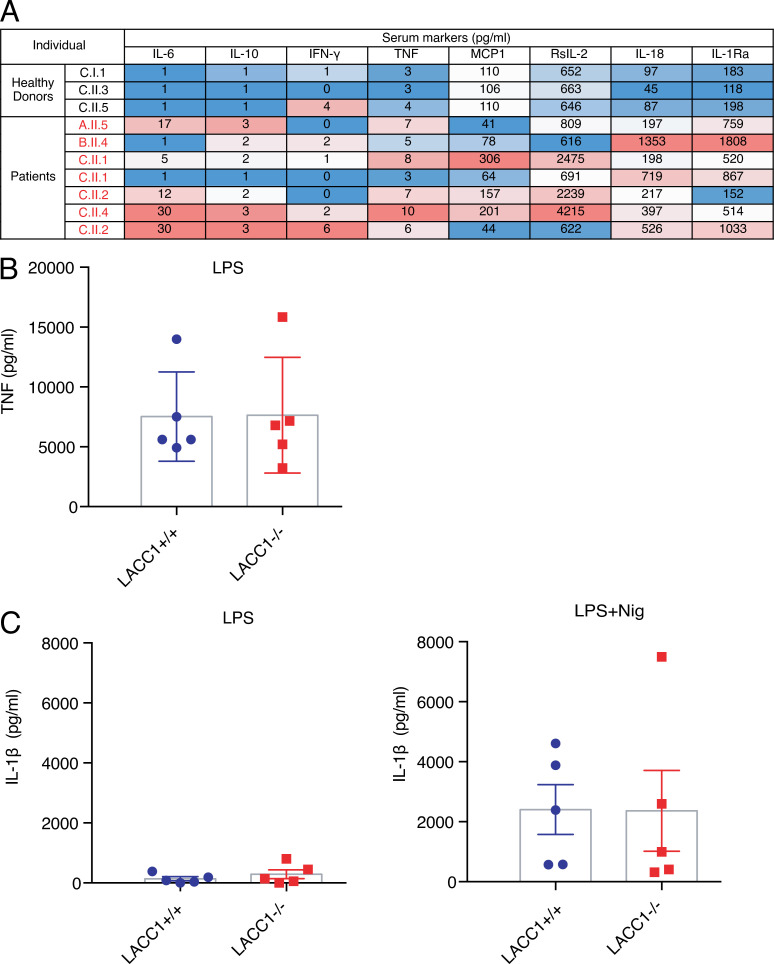
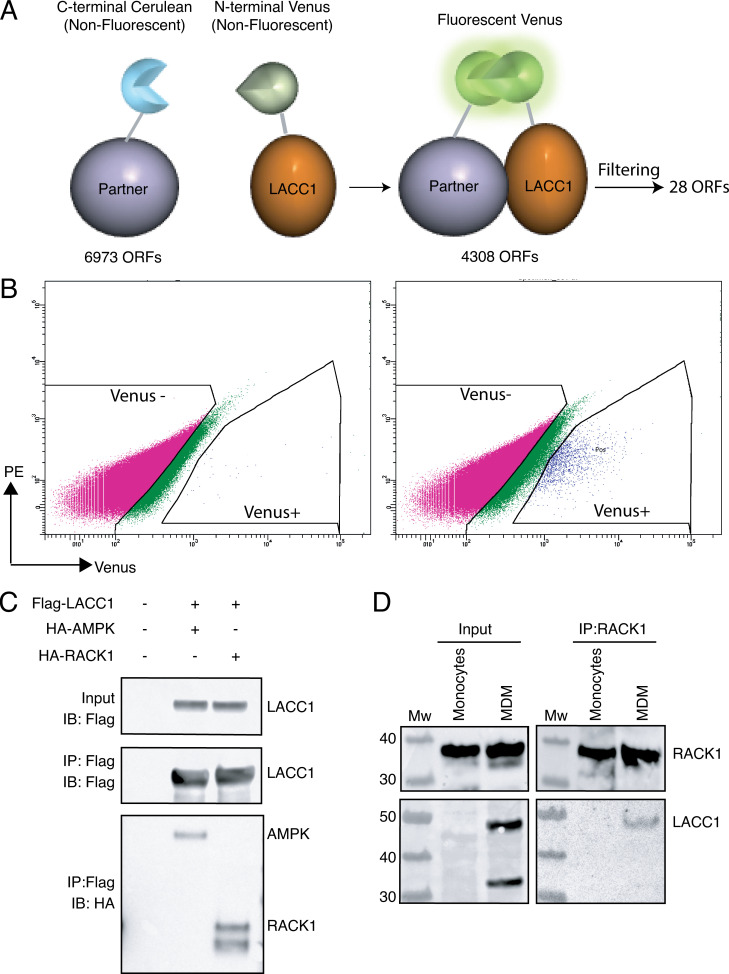
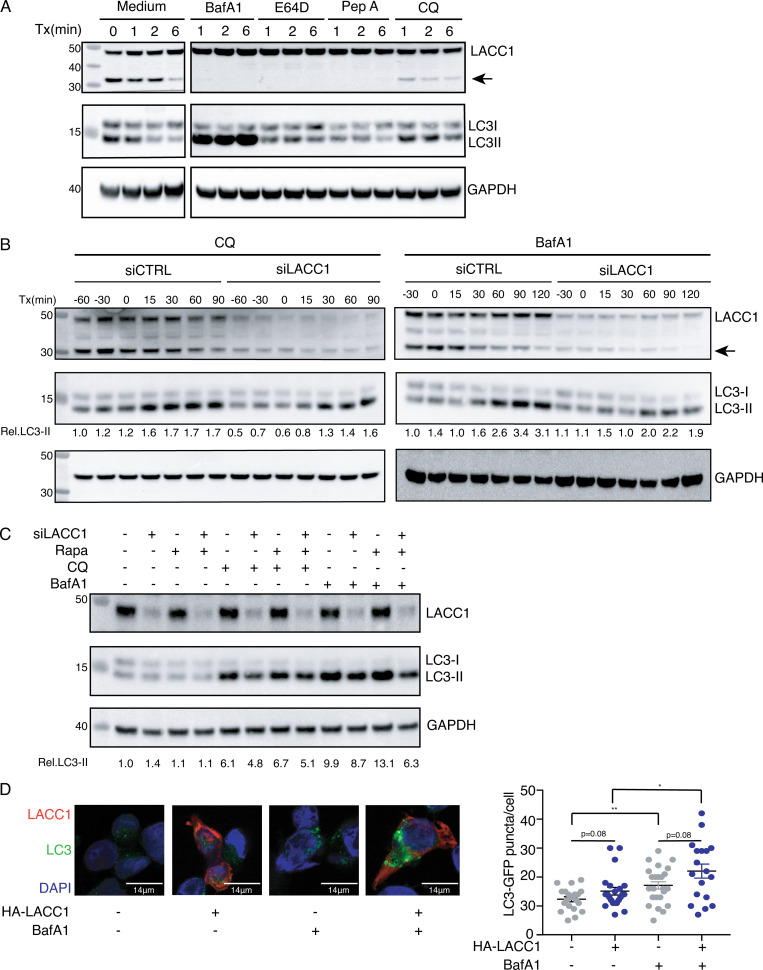
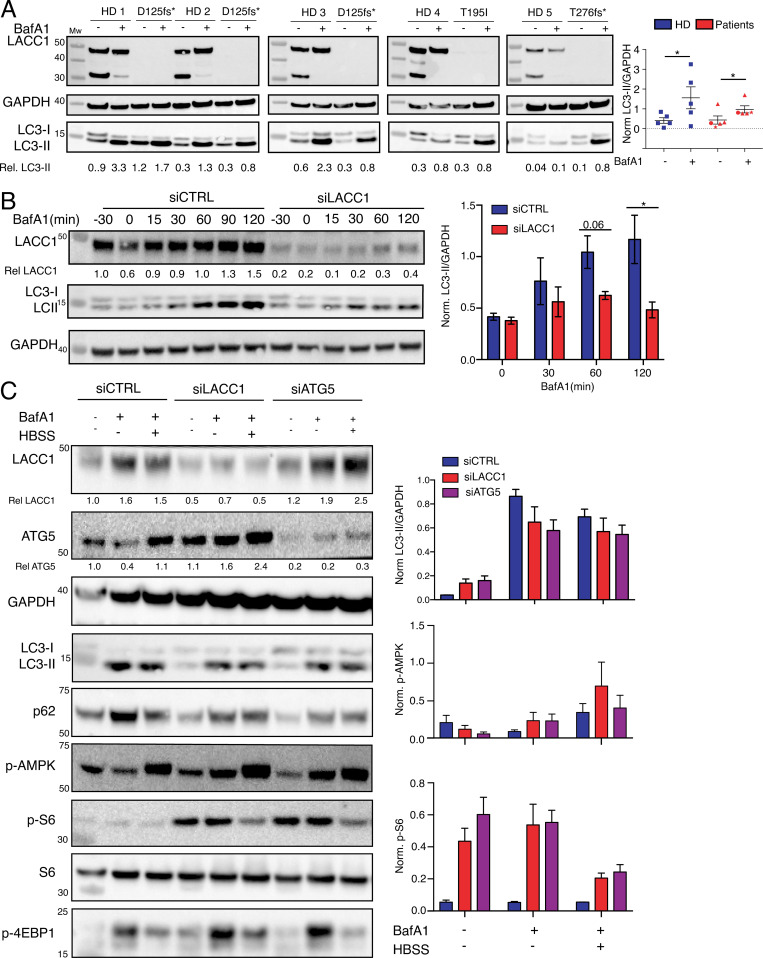
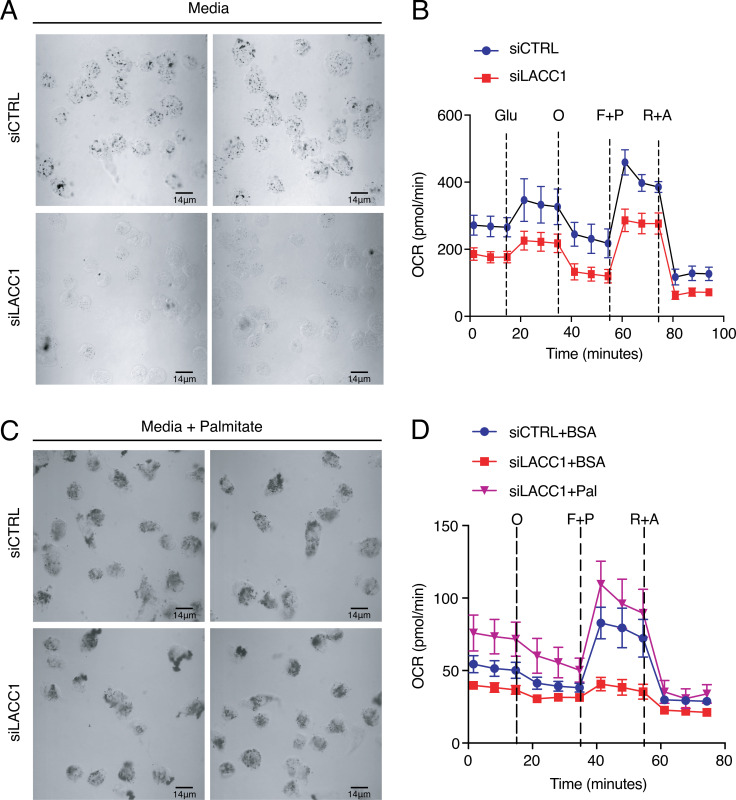
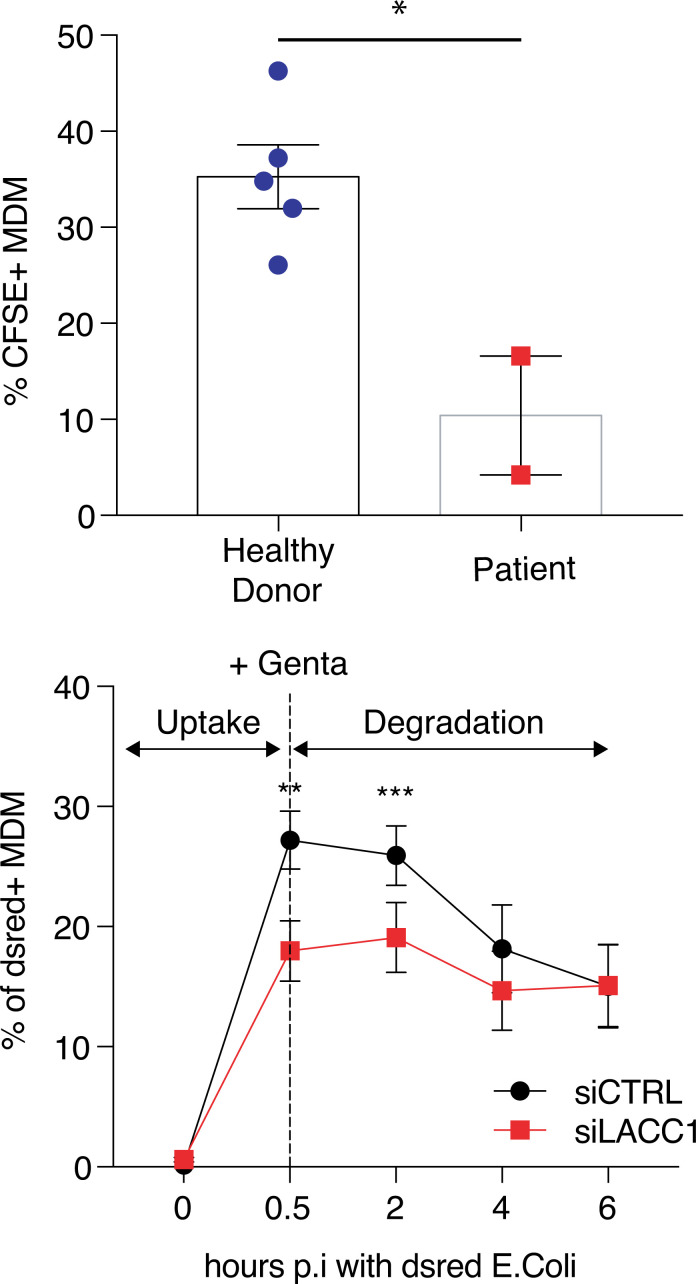
Juvenile idiopathic arthritis is the most common chronic rheumatic disease in children, and its etiology remains poorly understood. Here, we explored four families with early-onset arthritis carrying homozygous loss-of-expression mutations in LACC1. To understand the link between LACC1 and inflammation, we performed a functional study of LACC1 in human immune cells. We showed that LACC1 was primarily expressed in macrophages upon mTOR signaling. We found that LACC1 deficiency had no obvious impact on inflammasome activation, type I interferon response, or NF-κB regulation. Using bimolecular fluorescence complementation and biochemical assays, we showed that autophagy-inducing proteins, RACK1 and AMPK, interacted with LACC1. Autophagy blockade in macrophages was associated with LACC1 cleavage and degradation. Moreover, LACC1 deficiency reduced autophagy flux in primary macrophages. This was associated with a defect in the accumulation of lipid droplets and mitochondrial respiration, suggesting that LACC1-dependent autophagy fuels macrophage bioenergetics metabolism. Altogether, LACC1 deficiency defines a novel form of genetically inherited juvenile arthritis associated with impaired autophagy in macrophages.
© 2021 Omarjee et al.
Conflict of interest statement
Disclosures: S. Georgin-Lavialle reported personal fees from SOBI, non-financial support from Novartis, and personal fees from BMS outside the submitted work. F. Bleicher reported a patent to FR1655539 pending. J. Reboulet reported a patent to FR1655539 issued. S. Merabet reported a patent to FR1655539 pending. T. Henry reported personal fees from SOBI and grants from SOBI outside the submitted work. No other disclosures were reported.
Figures
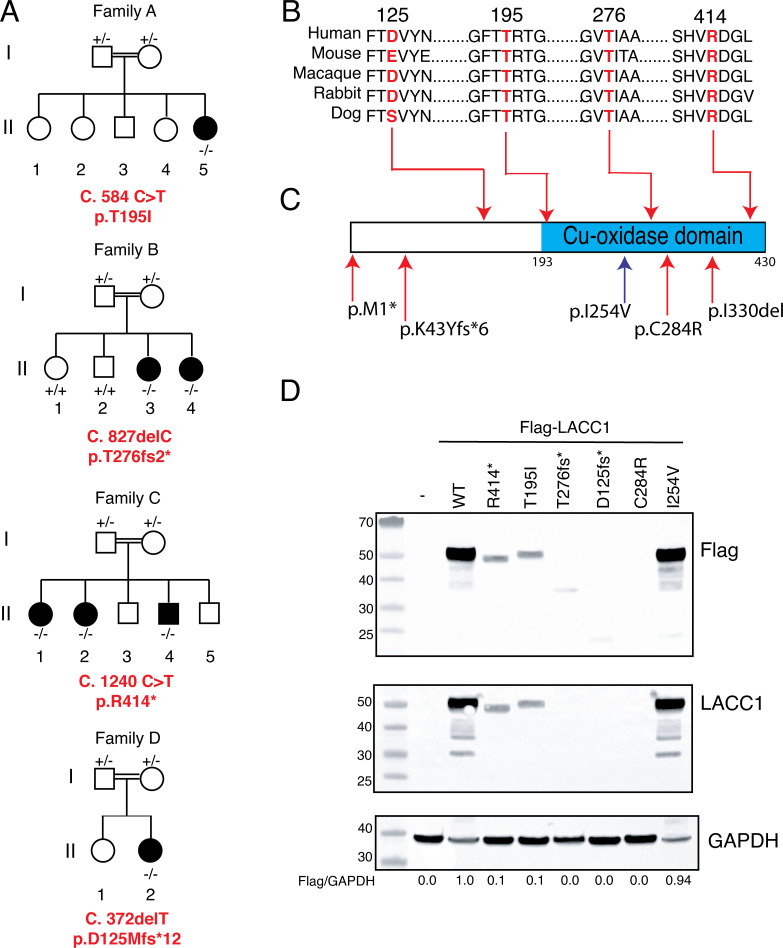
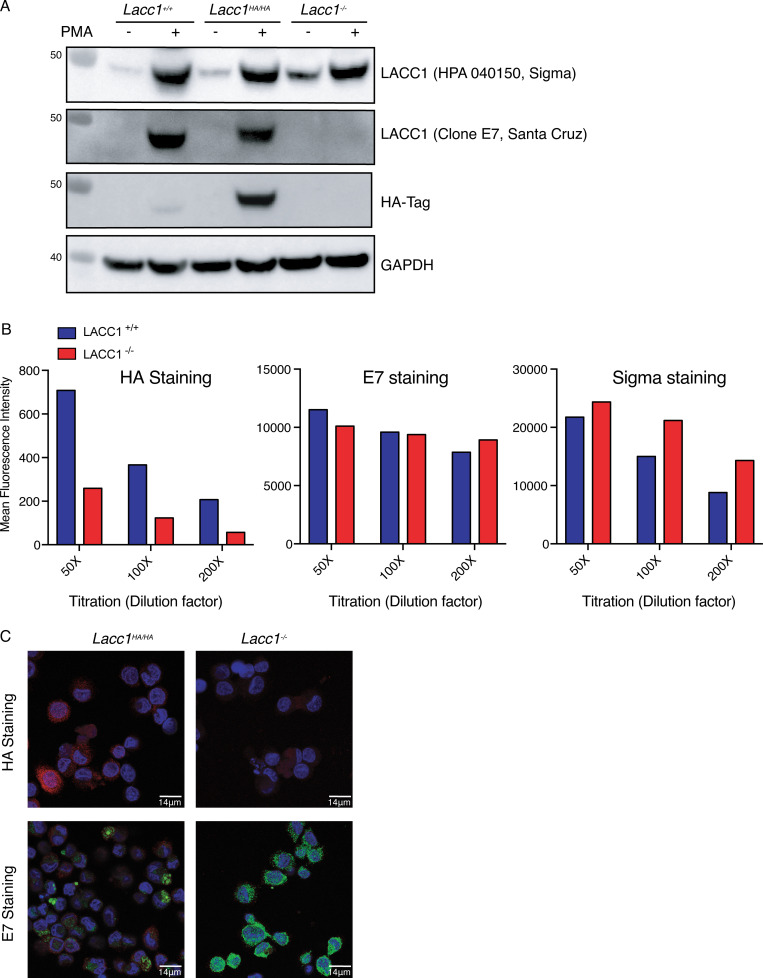
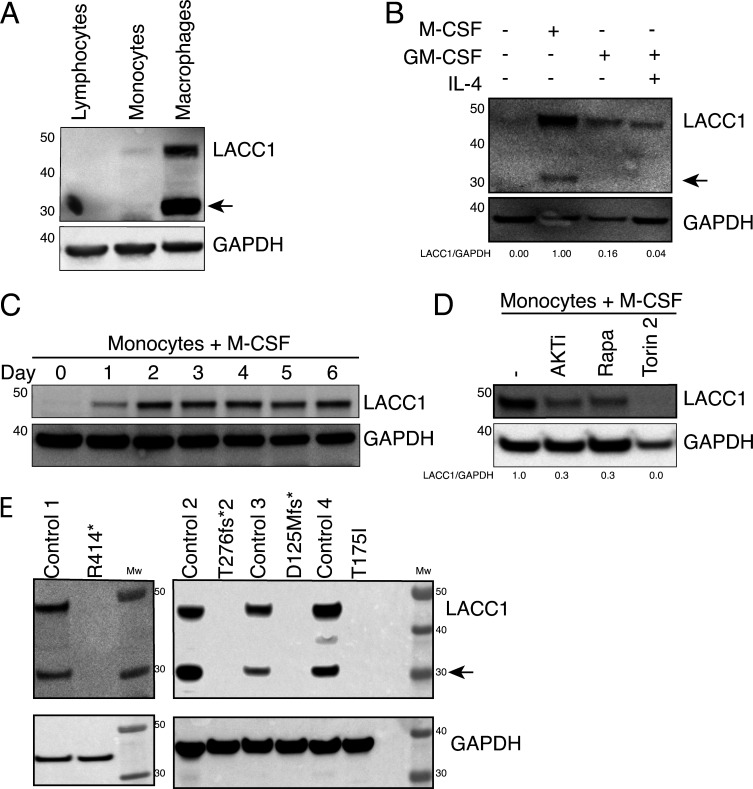
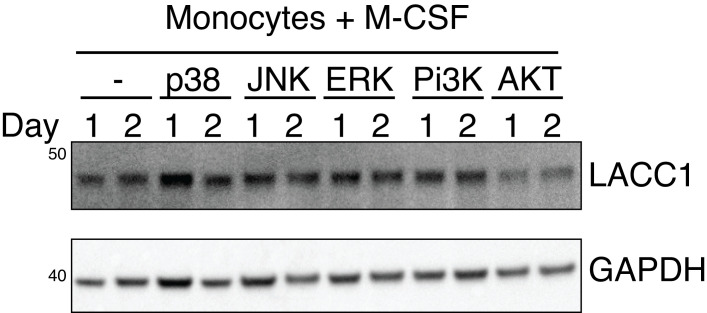
Comment in
-
LACC1-associated JIA linked to autophagy defects in macrophages.Nat Rev Rheumatol. 2021 May;17(5):251. doi: 10.1038/s41584-021-00607-0. Nat Rev Rheumatol. 2021. PMID: 33762710 No abstract available.
References
-
- Assadi, G., Saleh R., Hadizadeh F., Vesterlund L., Bonfiglio F., Halfvarson J., Törkvist L., Eriksson A.S., Harris H.E., Sundberg E., and D’Amato M.. 2016. LACC1 polymorphisms in inflammatory bowel disease and juvenile idiopathic arthritis. Genes Immun. 17:261–264. 10.1038/gene.2016.17 - DOI - PubMed
-
- Baldassano, R.N., Bradfield J.P., Monos D.S., Kim C.E., Glessner J.T., Casalunovo T., Frackelton E.C., Otieno F.G., Kanterakis S., Shaner J.L., et al. 2007. Association of the T300A non-synonymous variant of the ATG16L1 gene with susceptibility to paediatric Crohn’s disease. Gut. 56:1171–1173. 10.1136/gut.2007.122747 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
