

Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field
- PMID: 33606945
- PMCID: PMC8105287
- DOI: 10.1146/annurev-biophys-091720-102019
Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field
Abstract
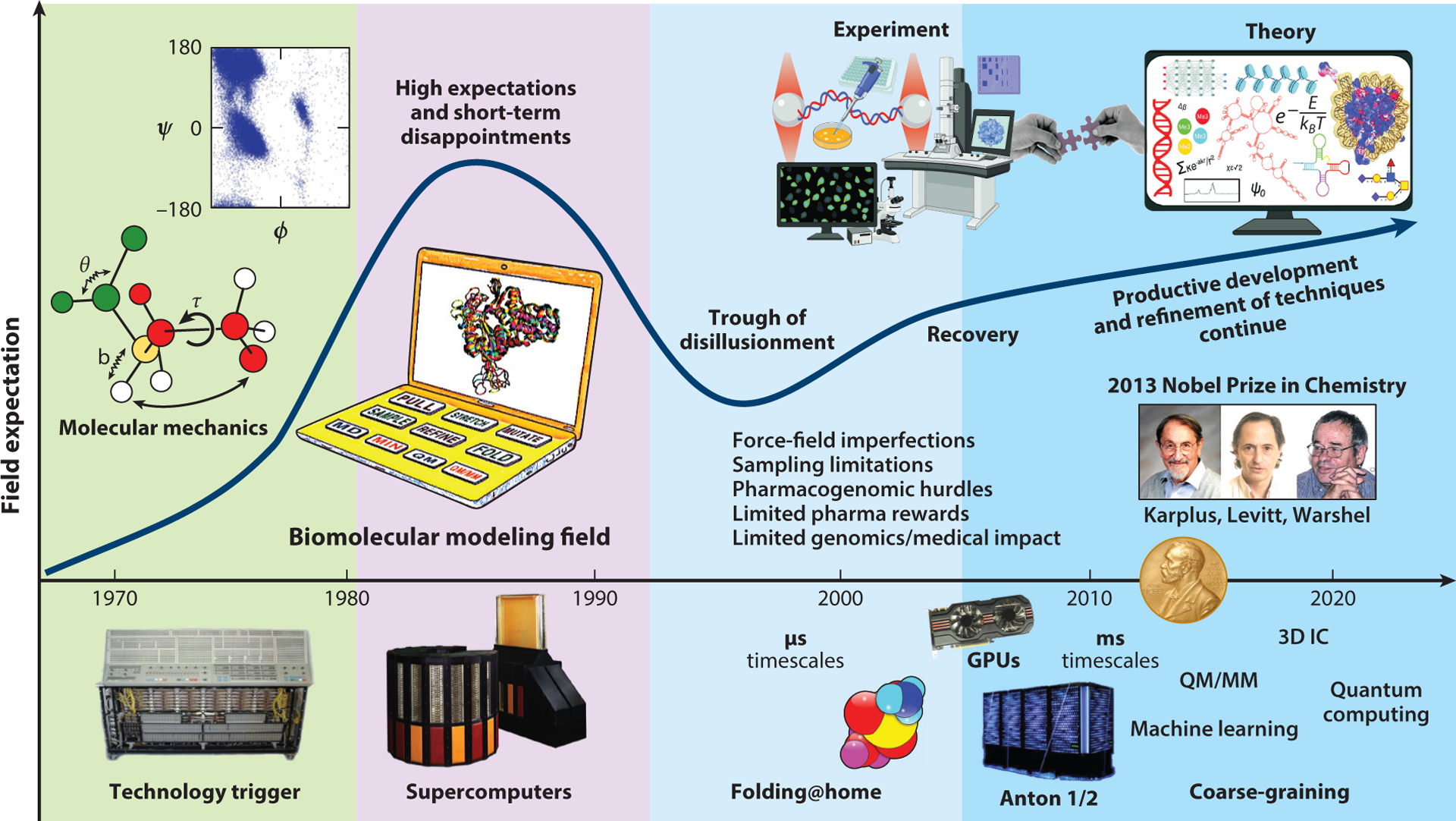
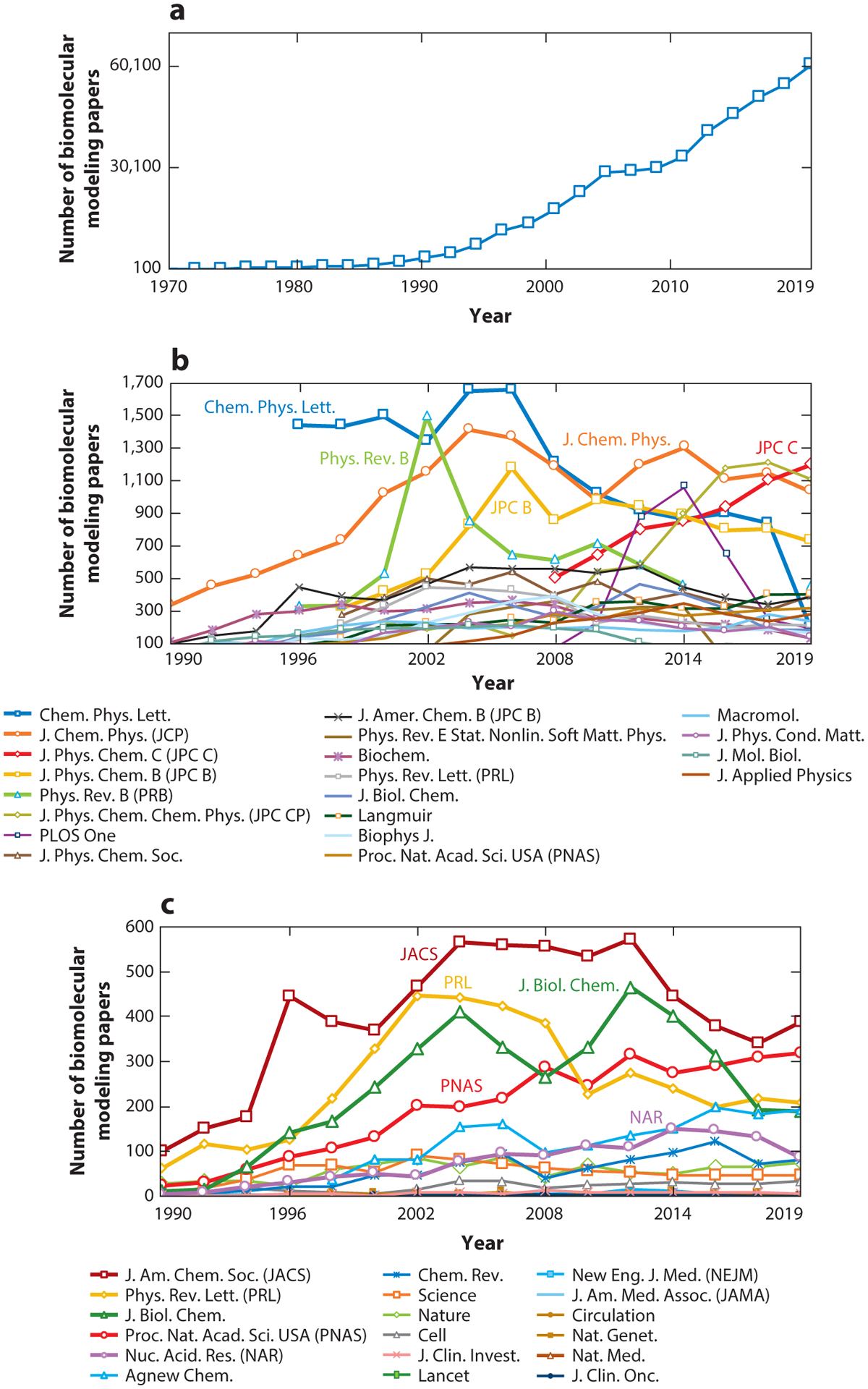
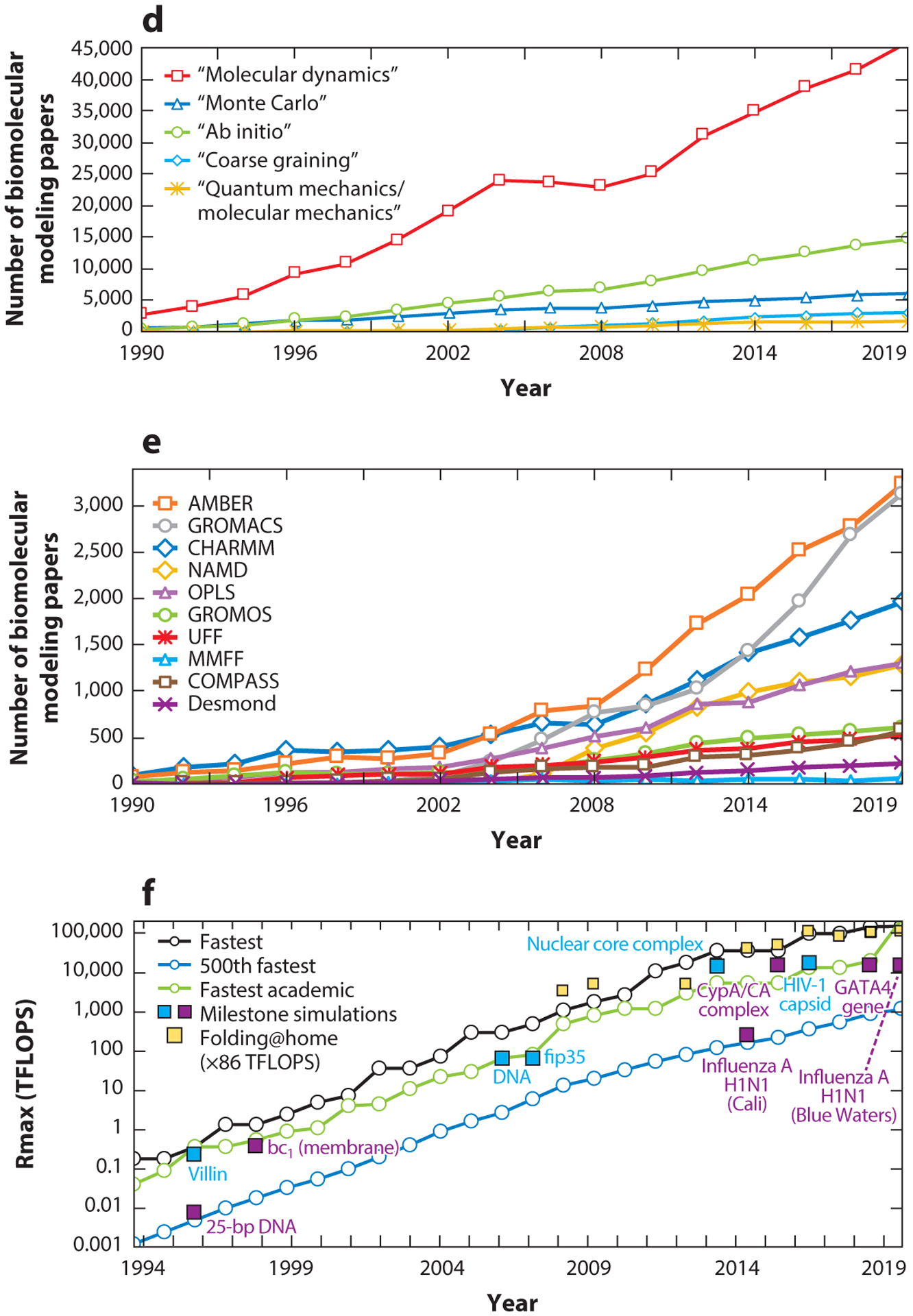
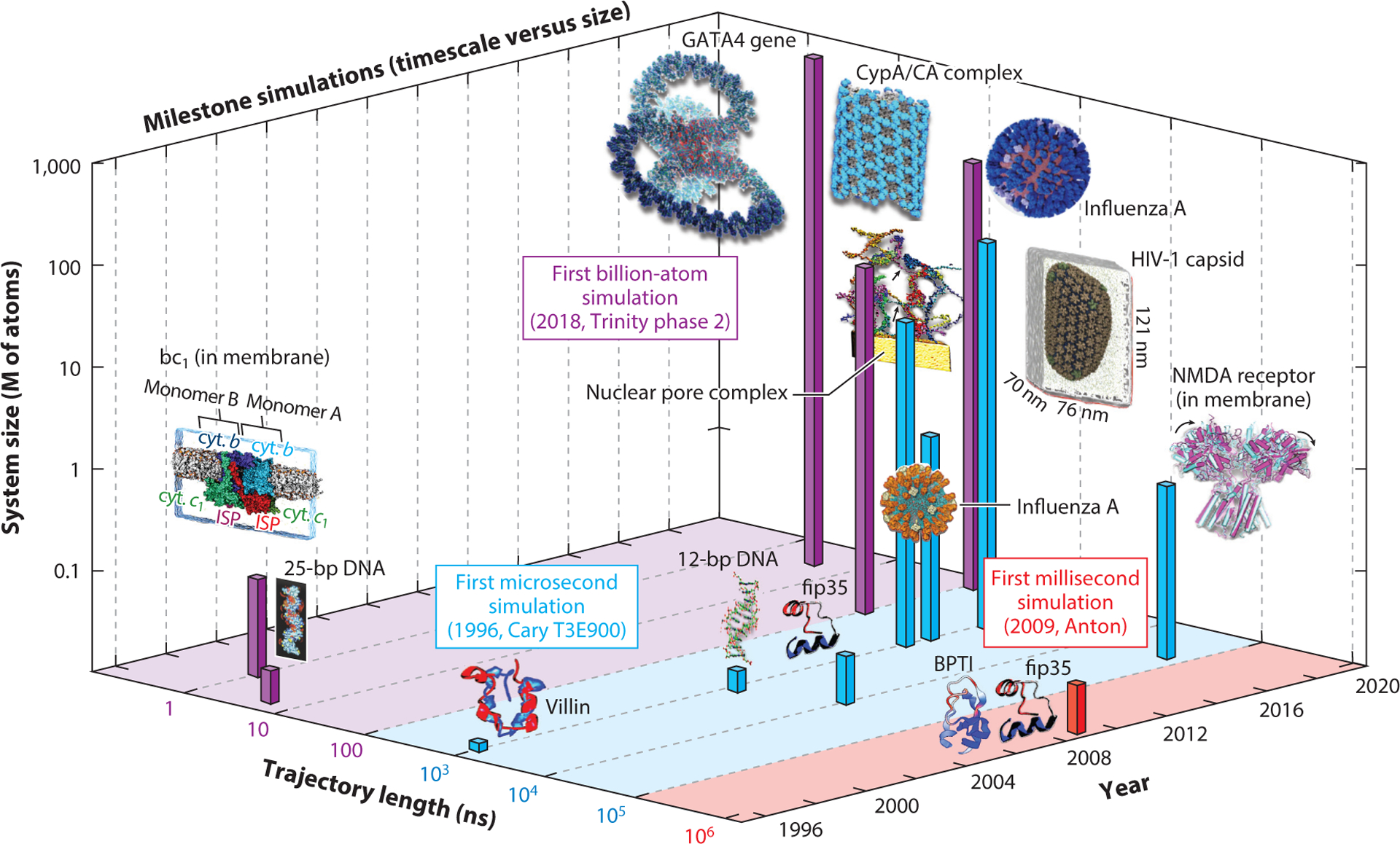
We reassess progress in the field of biomolecular modeling and simulation, following up on our perspective published in 2011. By reviewing metrics for the field's productivity and providing examples of success, we underscore the productive phase of the field, whose short-term expectations were overestimated and long-term effects underestimated. Such successes include prediction of structures and mechanisms; generation of new insights into biomolecular activity; and thriving collaborations between modeling and experimentation, including experiments driven by modeling. We also discuss the impact of field exercises and web games on the field's progress. Overall, we note tremendous success by the biomolecular modeling community in utilization of computer power; improvement in force fields; and development and application of new algorithms, notably machine learning and artificial intelligence. The combined advances are enhancing the accuracy andscope of modeling and simulation, establishing an exemplary discipline where experiment and theory or simulations are full partners.
Keywords: DNA folding; RNA folding; artificial intelligence; biomolecular dynamics; biomolecular modeling; biomolecular simulation; citizen science projects; machine learning; multiscale modeling; protein folding; structure prediction.
Figures
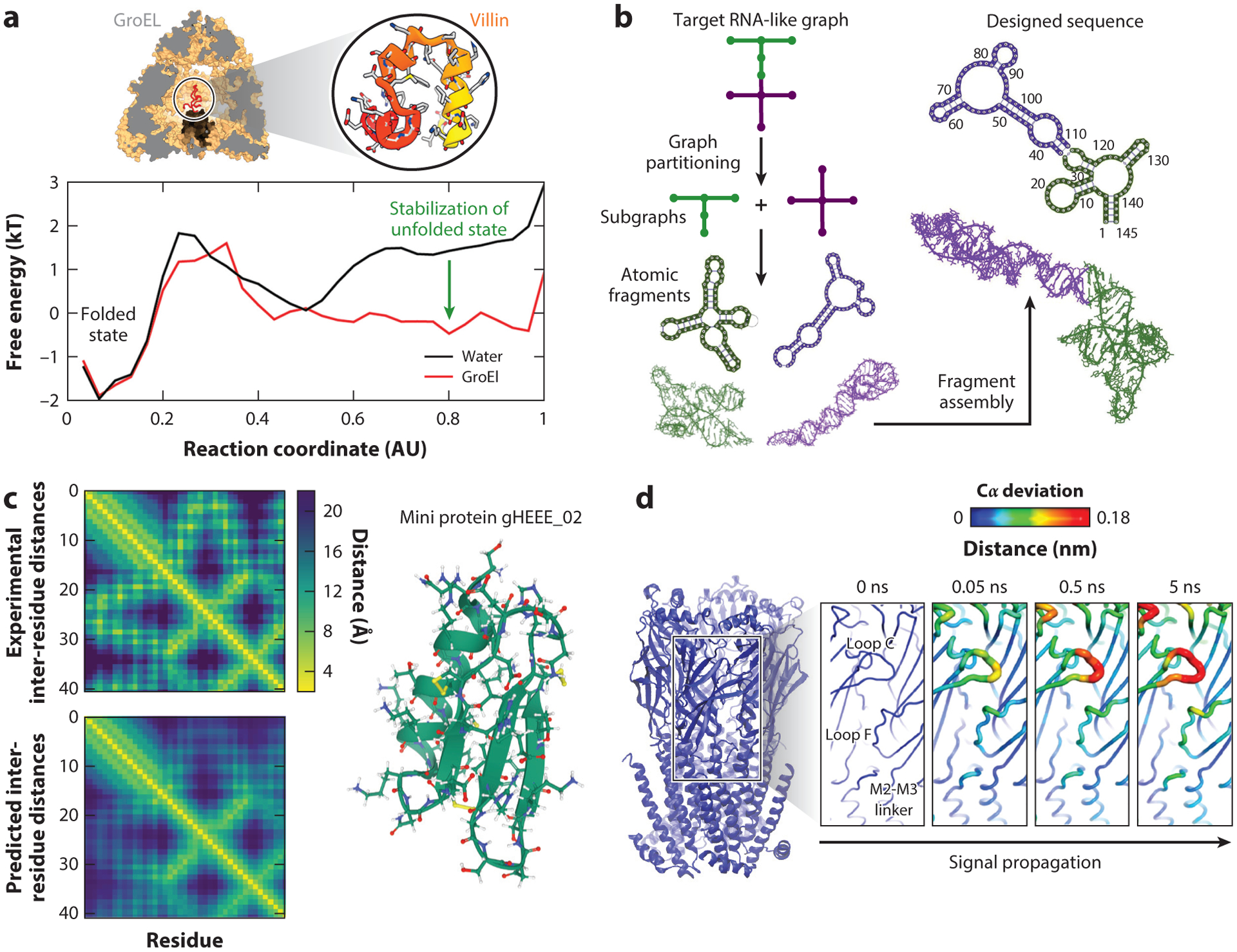
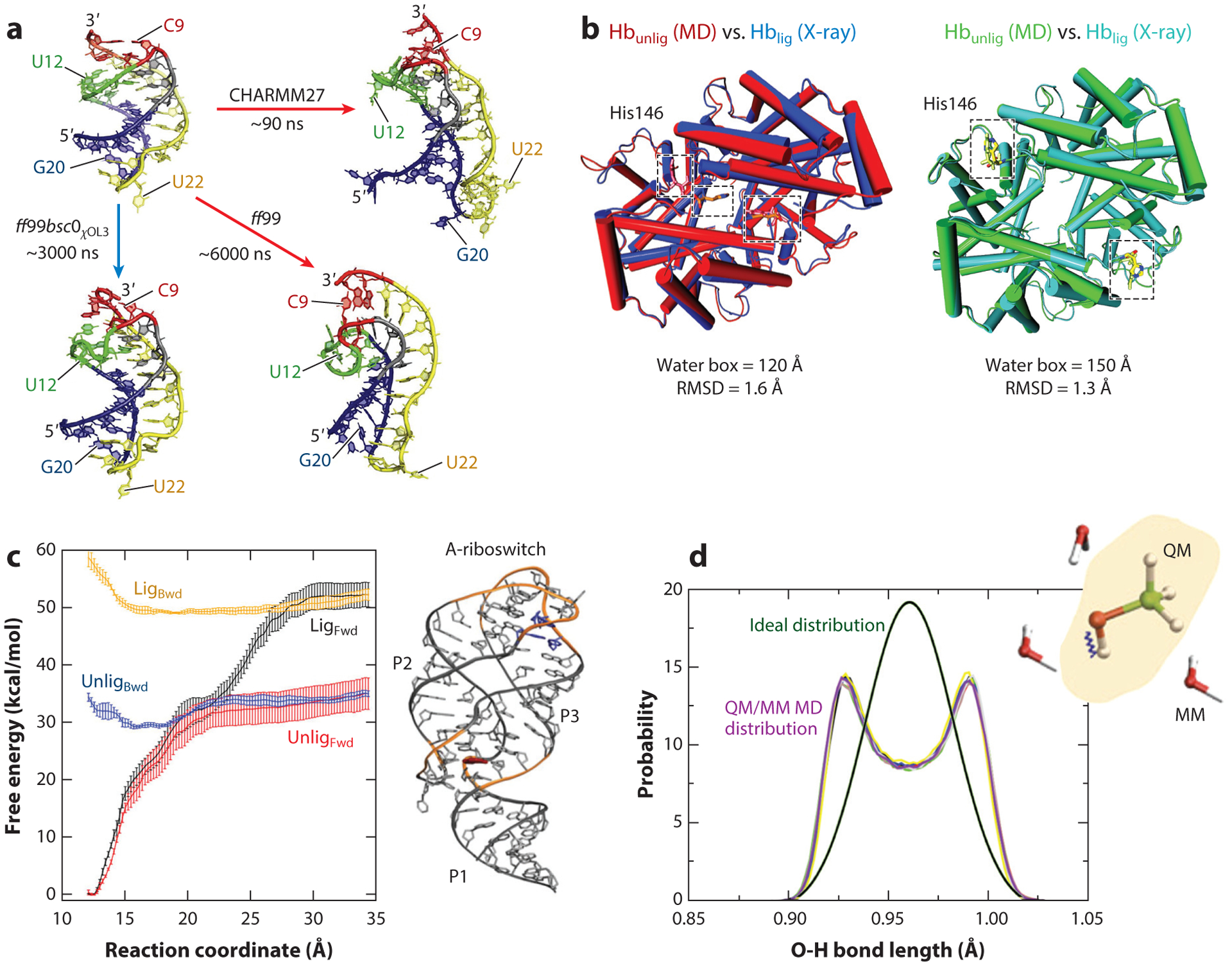
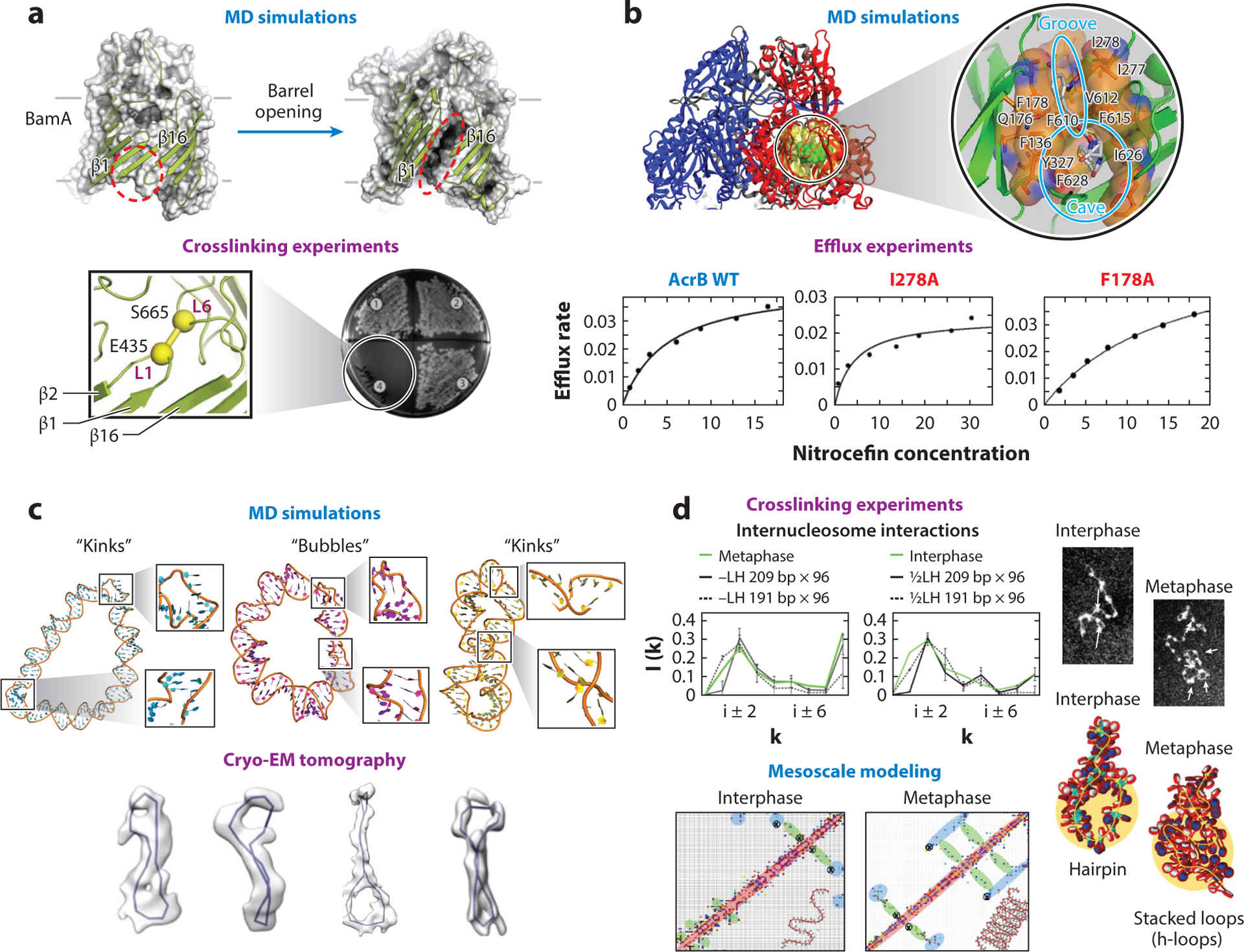
References
-
- Abriata LA, Tamo GE, Monastyrskyy B, Kryshtafovych A, Dal Peraro M. 2018. Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins 86(Suppl. 1):97–112 - PubMed
-
- Adamczak B, Kogut M, Czub J. 2018. Effect of osmolytes on the thermal stability of proteins: replica exchange simulations of Trp-cage in urea and betaine solutions. Phys. Chem. Chem. Phys 20(16):11174–82 - PubMed
-
- Alder BJ, Wainwright TE. 1959. Studies in molecular dynamics. I. General method. J. Chem. Phys 31(2):459–66
-
- Allinger NL. 1976. Calculation of molecular structure and energy by force-field methods. In Advances in Physical Organic Chemistry, Vol. 13, ed. Gold V, Bethell D, pp. 1–82. Cambridge, MA: Academic
-
- Allinger NL, Miller MA, Van Catledge FA, Hirsch JA. 1967. Conformational analysis. LVII. The calculation of the conformational structures of hydrocarbons by the Westheimer-Hendrickson-Wiberg method. J. Am. Chem. Soc 89(17):4345–57
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
