

Massive expansion of human gut bacteriophage diversity
- PMID: 33606979
- PMCID: PMC7895897
- DOI: 10.1016/j.cell.2021.01.029
Massive expansion of human gut bacteriophage diversity
Abstract
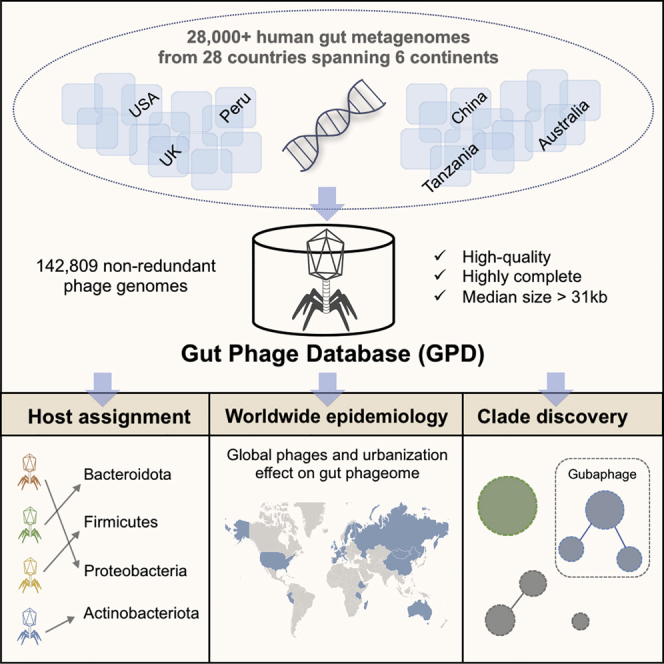
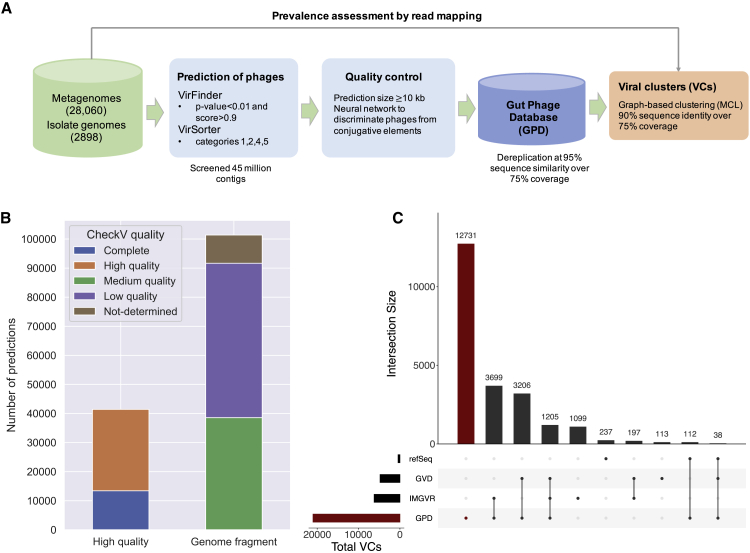
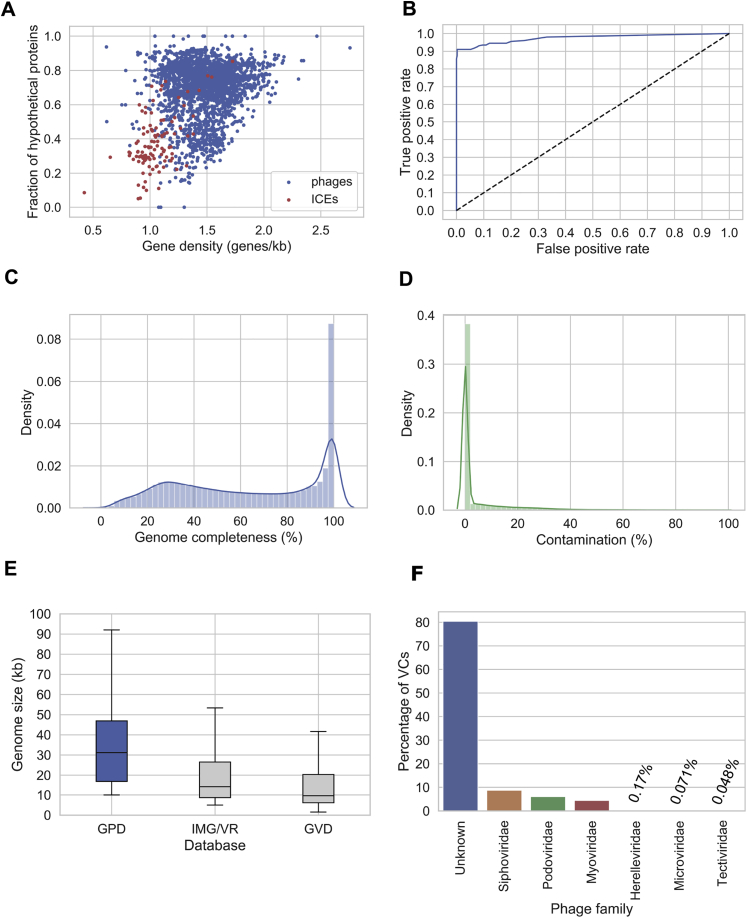
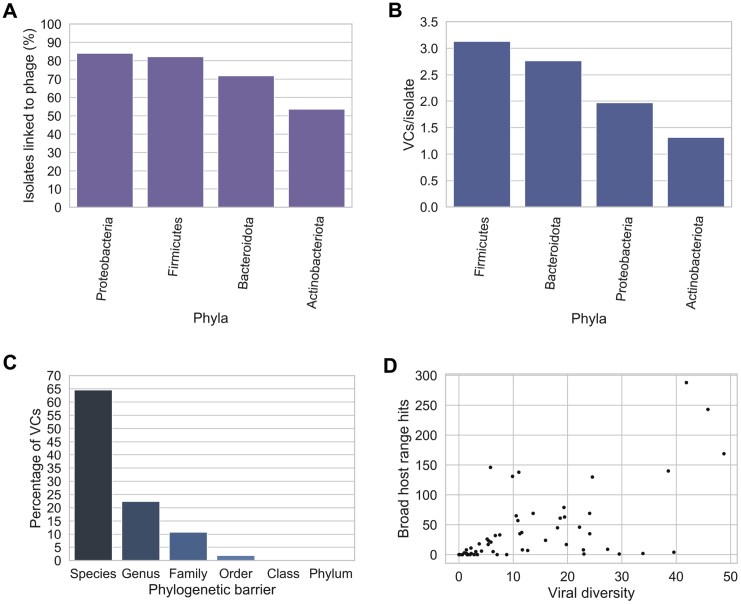
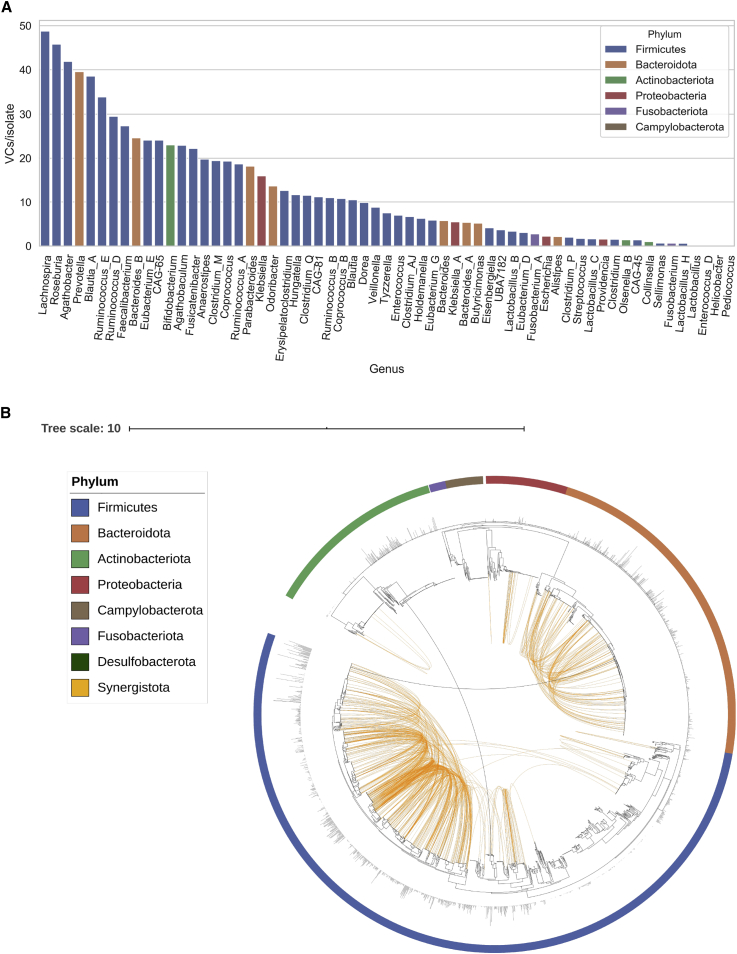
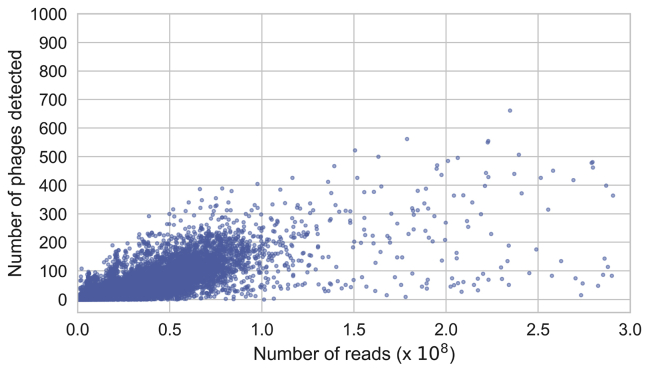
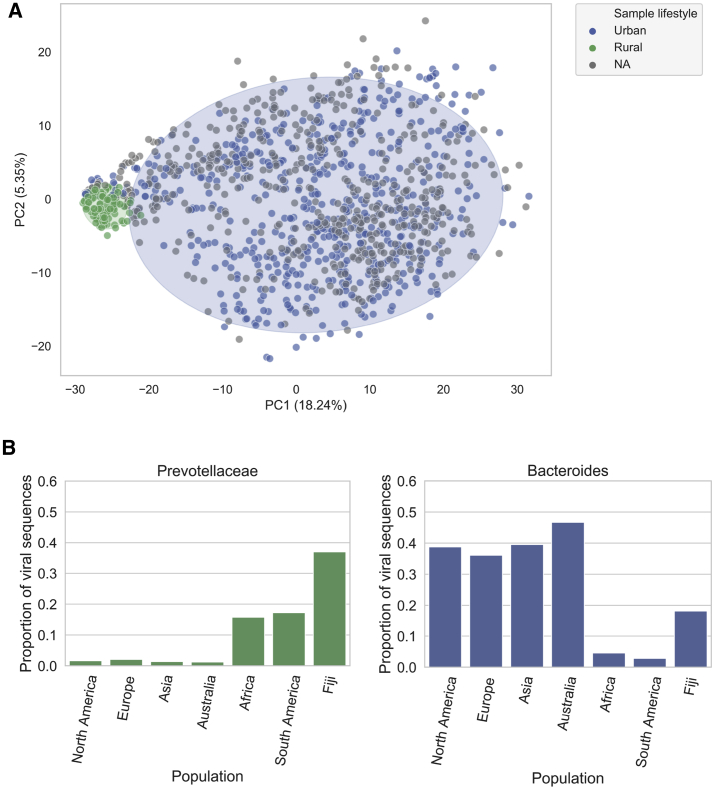
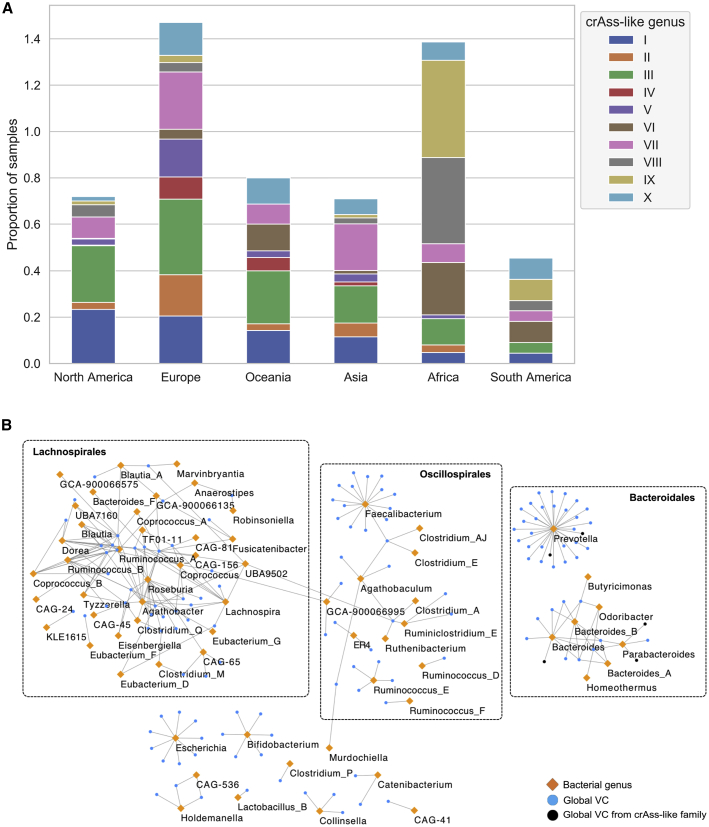
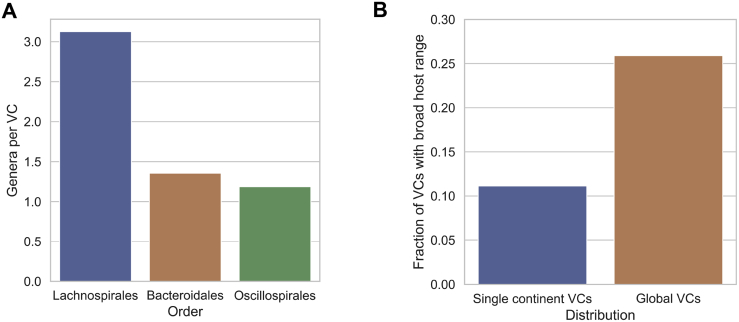
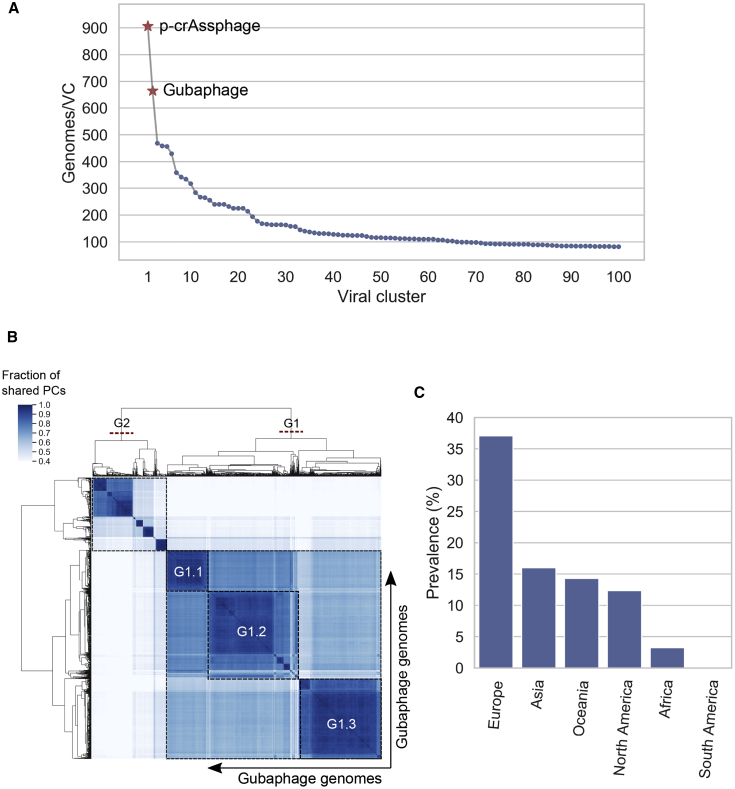
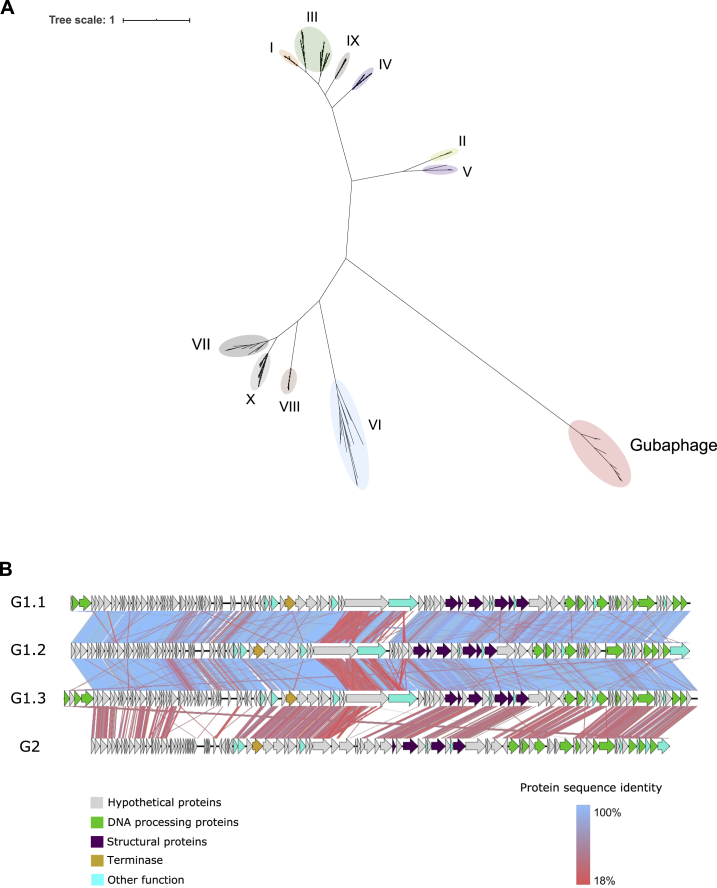
Bacteriophages drive evolutionary change in bacterial communities by creating gene flow networks that fuel ecological adaptions. However, the extent of viral diversity and its prevalence in the human gut remains largely unknown. Here, we introduce the Gut Phage Database, a collection of ∼142,000 non-redundant viral genomes (>10 kb) obtained by mining a dataset of 28,060 globally distributed human gut metagenomes and 2,898 reference genomes of cultured gut bacteria. Host assignment revealed that viral diversity is highest in the Firmicutes phyla and that ∼36% of viral clusters (VCs) are not restricted to a single species, creating gene flow networks across phylogenetically distinct bacterial species. Epidemiological analysis uncovered 280 globally distributed VCs found in at least 5 continents and a highly prevalent phage clade with features reminiscent of p-crAssphage. This high-quality, large-scale catalog of phage genomes will improve future virome studies and enable ecological and evolutionary analysis of human gut bacteriophages.
Keywords: database; gut bacteria; human gut; metagenomics; microbiome; phage; virus.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests T.D.L. is the co-founder and Chief Scientific Officer of Microbiotica Pty Ltd
Figures
References
-
- Abadi M., Barham P., Chen J., Chen Z., Davis A., Dean J., Devin M., Ghemawat S., Irving G., Isard M. Proceedings of the 12th USENIX Conference on Operating Systems Design and Implementation. USENIX Association; 2016. TensorFlow: a system for large-scale machine learning; pp. 265–283.
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
