

Patient-tailored design for selective co-inhibition of leukemic cell subpopulations
- PMID: 33608276
- PMCID: PMC7895436
- DOI: 10.1126/sciadv.abe4038
Patient-tailored design for selective co-inhibition of leukemic cell subpopulations
Abstract
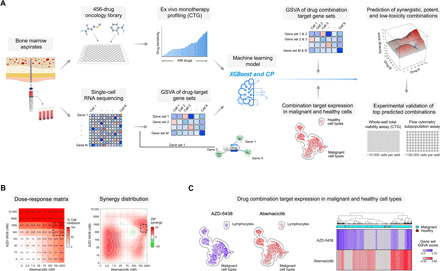
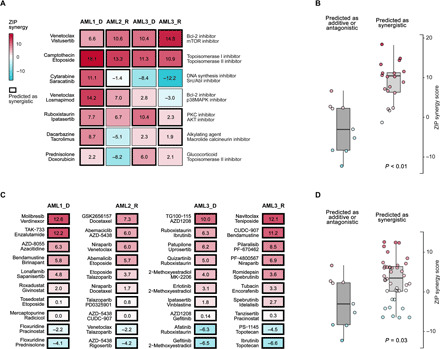
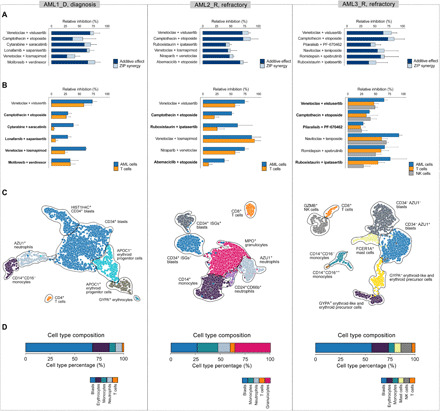
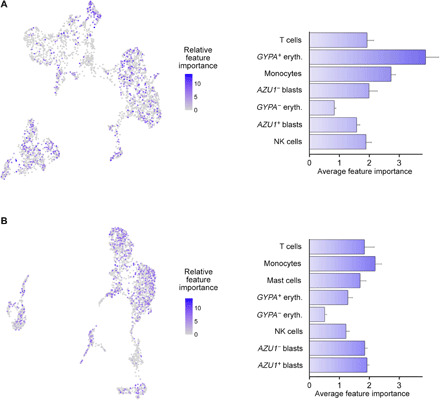
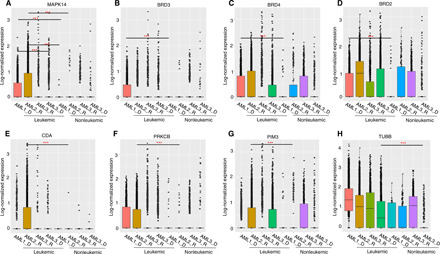
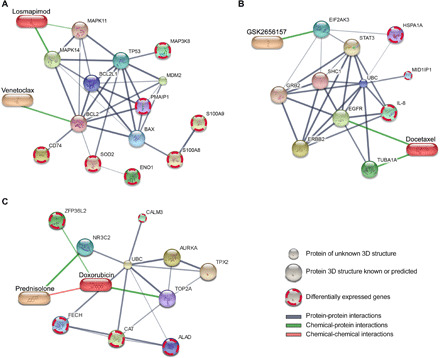
The extensive drug resistance requires rational approaches to design personalized combinatorial treatments that exploit patient-specific therapeutic vulnerabilities to selectively target disease-driving cell subpopulations. To solve the combinatorial explosion challenge, we implemented an effective machine learning approach that prioritizes patient-customized drug combinations with a desired synergy-efficacy-toxicity balance by combining single-cell RNA sequencing with ex vivo single-agent testing in scarce patient-derived primary cells. When applied to two diagnostic and two refractory acute myeloid leukemia (AML) patient cases, each with a different genetic background, we accurately predicted patient-specific combinations that not only resulted in synergistic cancer cell co-inhibition but also were capable of targeting specific AML cell subpopulations that emerge in differing stages of disease pathogenesis or treatment regimens. Our functional precision oncology approach provides an unbiased means for systematic identification of personalized combinatorial regimens that selectively co-inhibit leukemic cells while avoiding inhibition of nonmalignant cells, thereby increasing their likelihood for clinical translation.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Cancer Genome Atlas Research Network, Ley T. J., Miller C., Ding L., Raphael B. J., Mungall A. J., Robertson A. G., Hoadley K., Triche T. J. Jr., Laird P. W., Baty J. D., Fulton L. L., Fulton R., Heath S. E., Kalicki-Veizer J., Kandoth C., Klco J. M., Koboldt D. C., Kanchi K.-L., Kulkarni S., Lamprecht T. L., Larson D. E., Lin L., Lu C., McLellan M. D., McMichael J. F., Payton J., Schmidt H., Spencer D. H., Tomasson M. H., Wallis J. W., Wartman L. D., Watson M. A., Welch J., Wendl M. C., Ally A., Balasundaram M., Birol I., Butterfield Y., Chiu R., Chu A., Chuah E., Chun H.-J., Corbett R., Dhalla N., Guin R., He A., Hirst C., Hirst M., Holt R. A., Jones S., Karsan A., Lee D., Li H. I., Marra M. A., Mayo M., Moore R. A., Mungall K., Parker J., Pleasance E., Plettner P., Schein J., Stoll D., Swanson L., Tam A., Thiessen N., Varhol R., Wye N., Zhao Y., Gabriel S., Getz G., Sougnez C., Zou L., Leiserson M. D. M., Vandin F., Wu H.-T., Applebaum F., Baylin S. B., Akbani R., Broom B. M., Chen K., Motter T. C., Nguyen K., Weinstein J. N., Zhang N., Ferguson M. L., Adams C., Black A., Bowen J., Gastier-Foster J., Grossman T., Lichtenberg T., Wise L., Davidsen T., Demchok J. A., Shaw K. R. M., Sheth M., Sofia H. J., Yang L., Downing J. R., Eley G., Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013). - PMC - PubMed
-
- Quek L., David M. D., Kennedy A., Metzner M., Amatangelo M., Shih A., Stoilova B., Quivoron C., Heiblig M., Willekens C., Saada V., Alsafadi S., Vijayabaskar M. S., Peniket A., Bernard O. A., Agresta S., Yen K., Beth K. M., Stein E., Vassiliou G. S., Levine R., De Botton S., Thakurta A., Penard-Lacronique V., Vyas P., Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 24, 1167–1177 (2018). - PMC - PubMed
-
- Short N. J., Konopleva M., Kadia T. M., Borthakur G., Ravandi F., DiNardo C. D., Daver N., Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 10, 506–525 (2020). - PubMed
-
- Intlekofer A. M., Shih A. H., Wang B., Nazir A., Rustenburg A. S., Albanese S. K., Patel M., Famulare C., Correa F. M., Takemoto N., Durani V., Liu H., Taylor J., Farnoud N., Papaemmanuil E., Cross J. R., Tallman M. S., Arcila M. E., Roshal M., Petsko G. A., Wu B., Choe S., Konteatis Z. D., Biller S. A., Chodera J. D., Thompson C. B., Levine R. L., Stein E. M., Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018). - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
