

The molecular biology of FMRP: new insights into fragile X syndrome
- PMID: 33608673
- PMCID: PMC8094212
- DOI: 10.1038/s41583-021-00432-0
The molecular biology of FMRP: new insights into fragile X syndrome
Abstract
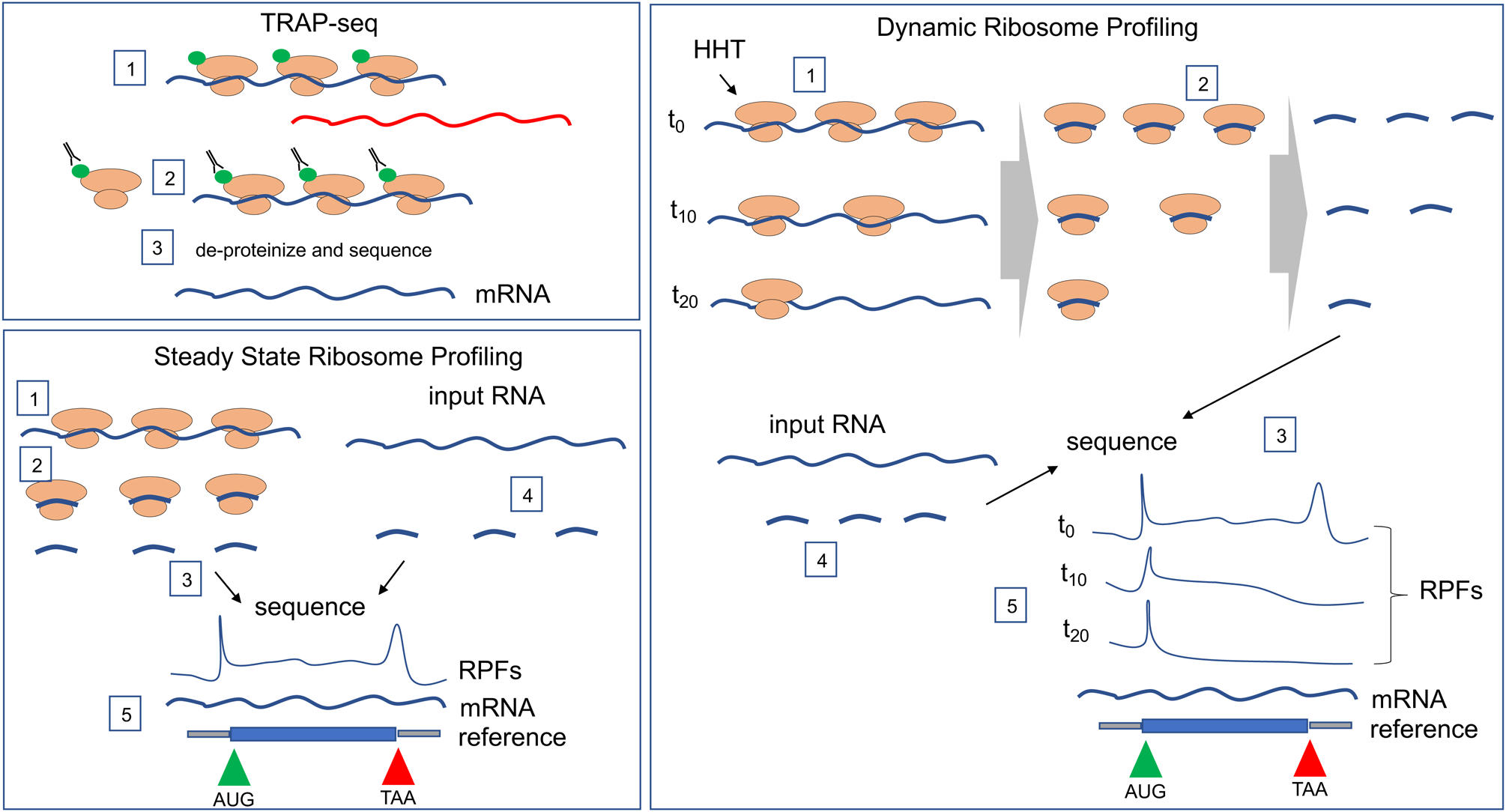
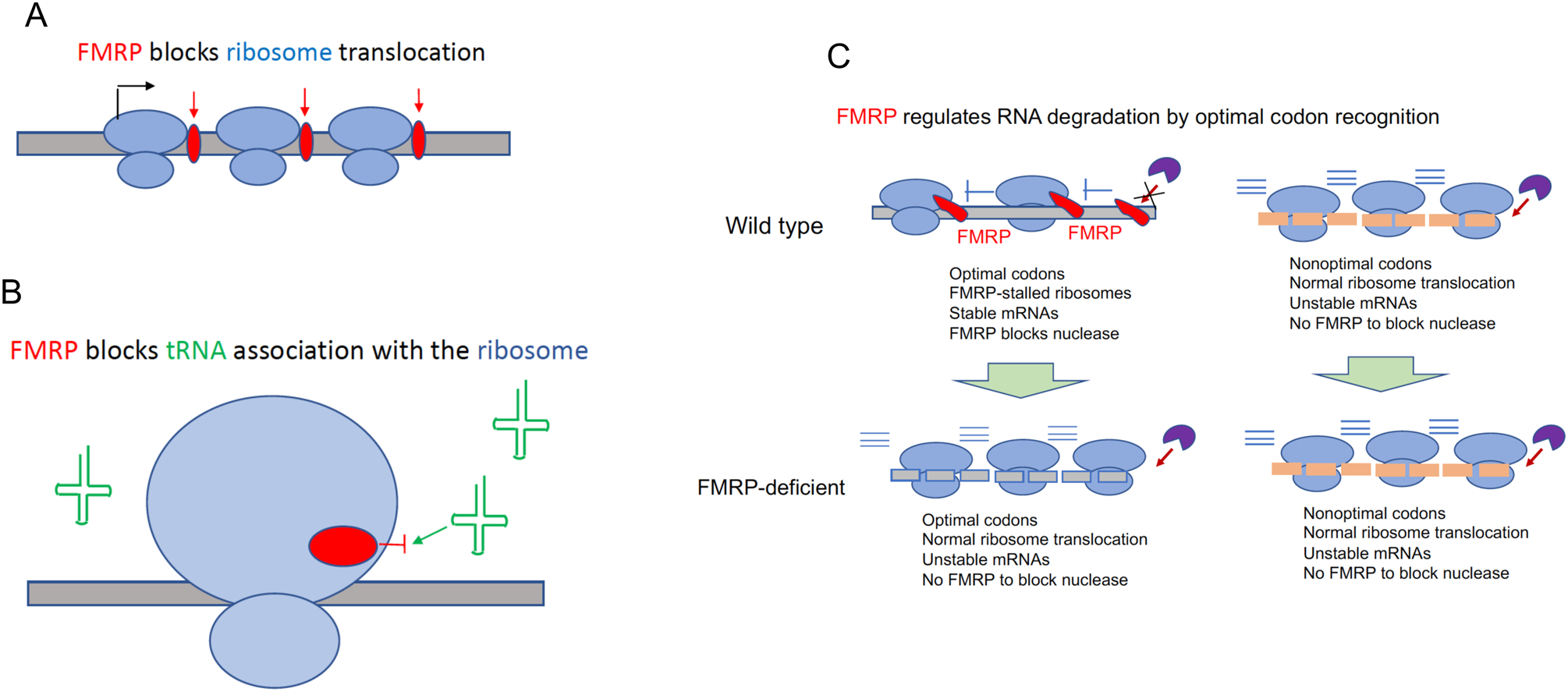
Fragile X mental retardation protein (FMRP) is the product of the fragile X mental retardation 1 gene (FMR1), a gene that - when epigenetically inactivated by a triplet nucleotide repeat expansion - causes the neurodevelopmental disorder fragile X syndrome (FXS). FMRP is a widely expressed RNA-binding protein with activity that is essential for proper synaptic plasticity and architecture, aspects of neural function that are known to go awry in FXS. Although the neurophysiology of FXS has been described in remarkable detail, research focusing on the molecular biology of FMRP has only scratched the surface. For more than two decades, FMRP has been well established as a translational repressor; however, recent whole transcriptome and translatome analyses in mouse and human models of FXS have shown that FMRP is involved in the regulation of nearly all aspects of gene expression. The emerging mechanistic details of the mechanisms by which FMRP regulates gene expression may offer ways to design new therapies for FXS.
Figures



References
-
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, & Nelson DL Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822 (1991). - PubMed
-
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, & Zhang FP Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). - PubMed
-
- Schaefer GB, & Mendelsohn NJ Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med 10, 4–12 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
