

Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease
- PMID: 33609480
- PMCID: PMC8285036
- DOI: 10.1016/S1474-4422(20)30477-4
Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease
Erratum in
-
Correction to Lancet Neurology 2021; 20: 235-46.Lancet Neurol. 2021 Apr;20(4):e3. doi: 10.1016/S1474-4422(21)00069-7. Epub 2021 Feb 24. Lancet Neurol. 2021. PMID: 33639092 No abstract available.
Abstract
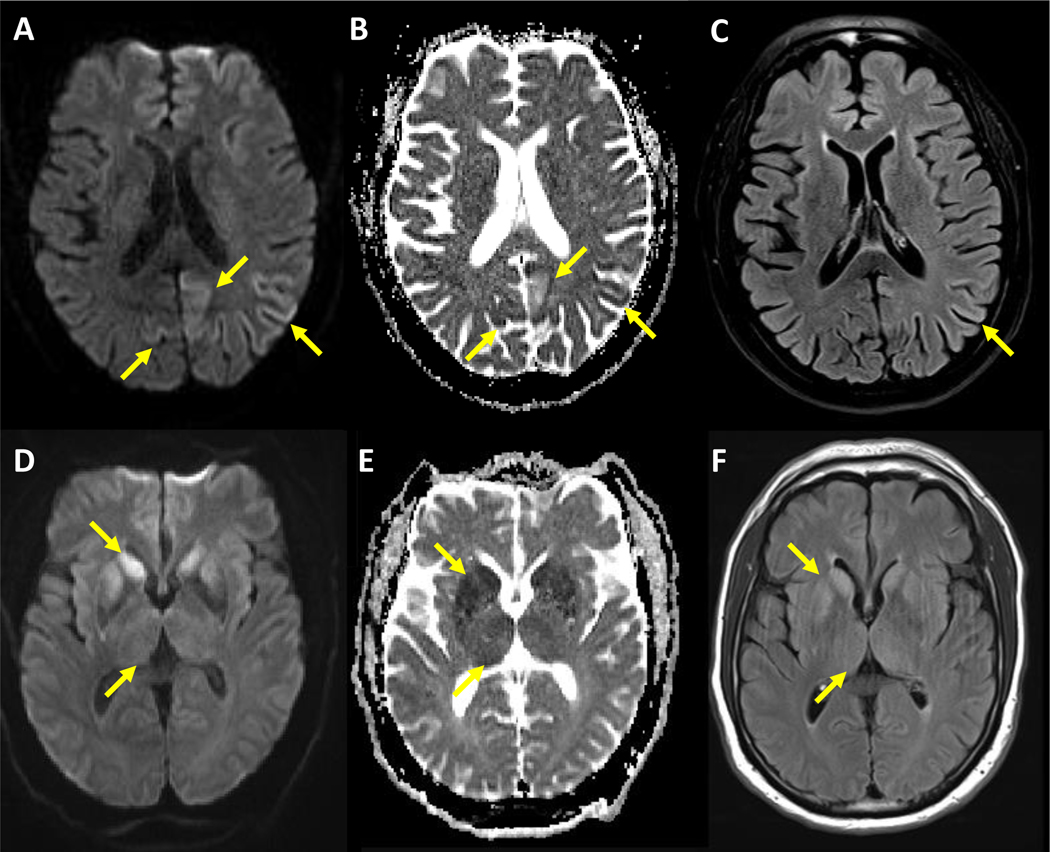
Sporadic Creutzfeldt-Jakob disease is a fatal neurodegenerative disease caused by misfolded prion proteins (PrPSc). Effective therapeutics are currently not available and accurate diagnosis can be challenging. Clinical diagnostic criteria use a combination of characteristic neuropsychiatric symptoms, CSF proteins 14-3-3, MRI, and EEG. Supportive biomarkers, such as high CSF total tau, could aid the diagnostic process. However, discordant studies have led to controversies about the clinical value of some established surrogate biomarkers. Development and clinical application of disease-specific protein aggregation and amplification assays, such as real-time quaking induced conversion (RT-QuIC), have constituted major breakthroughs for the confident pre-mortem diagnosis of sporadic Creutzfeldt-Jakob disease. Updated criteria for the diagnosis of sporadic Creutzfeldt-Jakob disease, including application of RT-QuIC, should improve early clinical confirmation, surveillance, assessment of PrPSc seeding activity in different tissues, and trial monitoring. Moreover, emerging blood-based, prognostic, and potentially pre-symptomatic biomarker candidates are under investigation.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Figures

References
-
- Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005; 64: 1586–91. - PubMed
-
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46: 224–33. - PubMed
-
- WHO. Global Surveillance, diagnosis, and Therapy of Human Transmissible spongiform Encephalopathies: Report of WHO consultation, February 9–11, 1998, Geneva, Switzerland.
-
- Otto M, Wiltfang J, Cepek L, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2002; 58: 192–7 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
