

FLCN regulates transferrin receptor 1 transport and iron homeostasis
- PMID: 33609526
- PMCID: PMC7995610
- DOI: 10.1016/j.jbc.2021.100426
FLCN regulates transferrin receptor 1 transport and iron homeostasis
Abstract
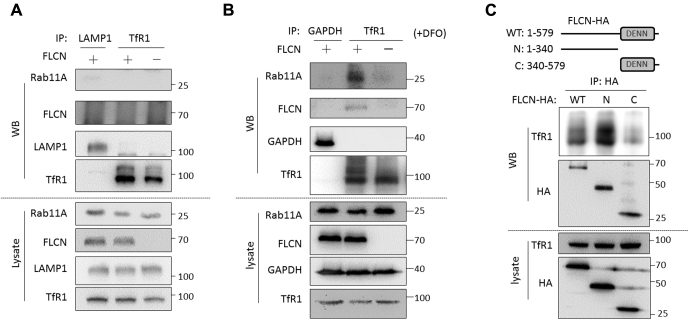
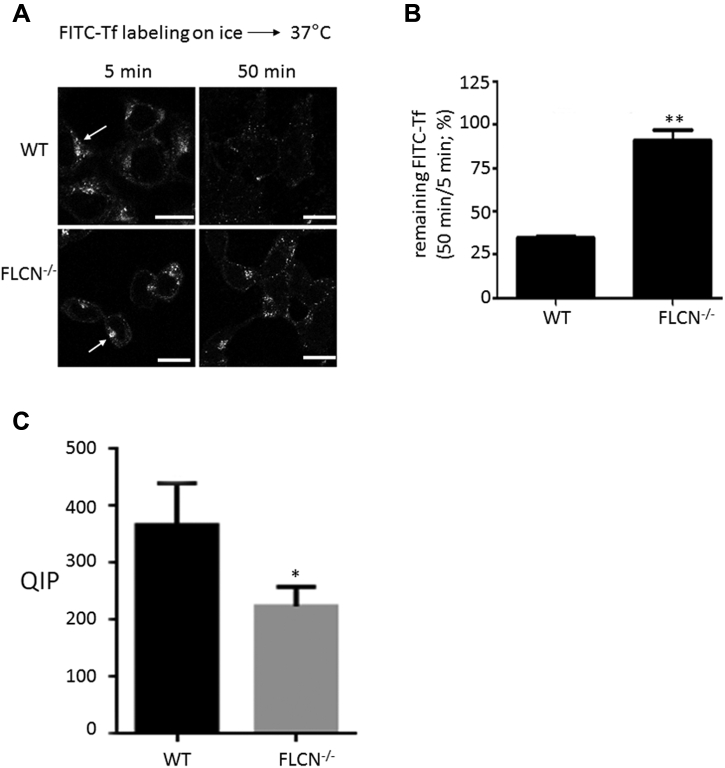
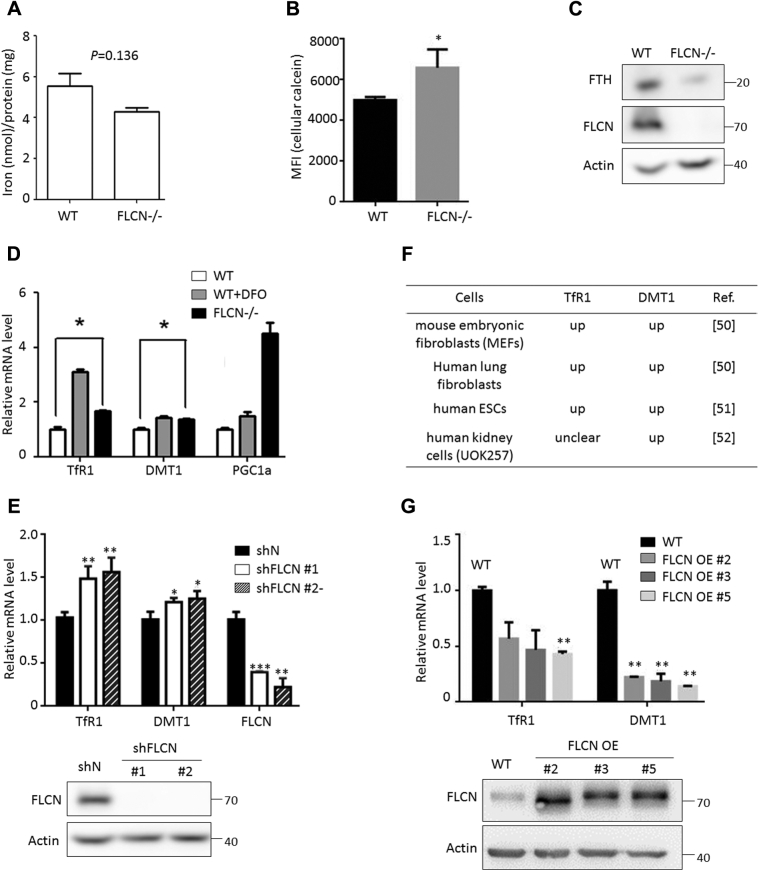
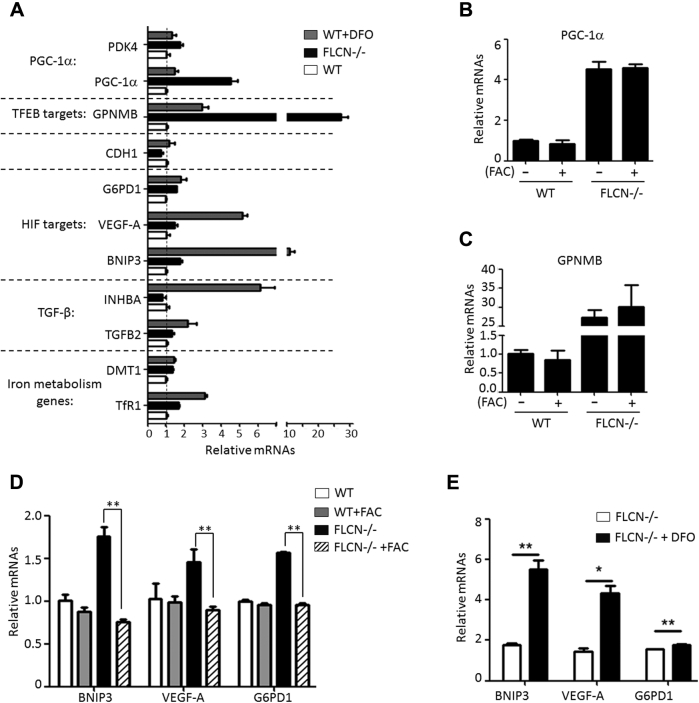
Birt-Hogg-Dubé (BHD) syndrome is a multiorgan disorder caused by inactivation of the folliculin (FLCN) protein. Previously, we identified FLCN as a binding protein of Rab11A, a key regulator of the endocytic recycling pathway. This finding implies that the abnormal localization of specific proteins whose transport requires the FLCN-Rab11A complex may contribute to BHD. Here, we used human kidney-derived HEK293 cells as a model, and we report that FLCN promotes the binding of Rab11A with transferrin receptor 1 (TfR1), which is required for iron uptake through continuous trafficking between the cell surface and the cytoplasm. Loss of FLCN attenuated the Rab11A-TfR1 interaction, resulting in delayed recycling transport of TfR1. This delay caused an iron deficiency condition that induced hypoxia-inducible factor (HIF) activity, which was reversed by iron supplementation. In a Drosophila model of BHD syndrome, we further demonstrated that the phenotype of BHD mutant larvae was substantially rescued by an iron-rich diet. These findings reveal a conserved function of FLCN in iron metabolism and may help to elucidate the mechanisms driving BHD syndrome.
Keywords: BHD; FLCN; HIF; iron.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures


Similar articles
-
FLCN is a novel Rab11A-interacting protein that is involved in the Rab11A-mediated recycling transport.J Cell Sci. 2018 Dec 19;131(24):jcs218792. doi: 10.1242/jcs.218792. J Cell Sci. 2018. PMID: 30446510
-
Birt-Hogg-Dube syndrome is a novel ciliopathy.Hum Mol Genet. 2013 Nov 1;22(21):4383-97. doi: 10.1093/hmg/ddt288. Epub 2013 Jun 19. Hum Mol Genet. 2013. PMID: 23784378 Free PMC article.
-
Birt-Hogg-Dubé: tumour suppressor function and signalling dynamics central to folliculin.Fam Cancer. 2013 Sep;12(3):367-72. doi: 10.1007/s10689-012-9576-9. Fam Cancer. 2013. PMID: 23096221
-
Birt-Hogg-Dubé syndrome: Clinical and molecular aspects of recently identified kidney cancer syndrome.Int J Urol. 2016 Mar;23(3):204-10. doi: 10.1111/iju.13015. Epub 2015 Nov 25. Int J Urol. 2016. PMID: 26608100 Review.
-
Focus on the pulmonary involvement and genetic patterns in Birt-Hogg-Dubè syndrome: Literature review.Respir Med. 2020 Jul;168:105995. doi: 10.1016/j.rmed.2020.105995. Epub 2020 May 6. Respir Med. 2020. PMID: 32469710 Review.
Cited by
-
Insights into the Molecular Mechanisms of Bushen Huoxue Decoction in Breast Cancer via Network Pharmacology and in vitro experiments.Curr Comput Aided Drug Des. 2025;21(1):50-66. doi: 10.2174/0115734099269728231115060827. Curr Comput Aided Drug Des. 2025. PMID: 39651565
References
-
- Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L., Duray P., Merino M., Choyke P., Pavlovich C.P., Sharma N., Walther M., Munroe D., Hill R., Maher E. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–164. - PubMed
-
- Baba M., Furihata M., Hong S.B., Tessarollo L., Haines D.C., Southon E., Patel V., Igarashi P., Alvord W.G., Leighty R., Yao M., Bernardo M., Ileva L., Choyke P., Warren M.B. Kidney-targeted Birt-Hogg-Dubé, gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 2008;100:140–154. - PMC - PubMed
-
- Gijezen L.M., Vernooij M., Martens H., Oduber C.E., Henquet C.J., Starink T.M., Prins M.H., Menko F.H., Nelemans P.J., van Steensel M.A. Topical rapamycin as a treatment for fibrofolliculomas in Birt-Hogg-Dubé syndrome: A double-blind placebo-controlled randomized split-face trial. PLoS One. 2014;9 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
