

Coronary Endothelium No-Reflow Injury Is Associated with ROS-Modified Mitochondrial Fission through the JNK-Drp1 Signaling Pathway
- PMID: 33613824
- PMCID: PMC7878075
- DOI: 10.1155/2021/6699516
Coronary Endothelium No-Reflow Injury Is Associated with ROS-Modified Mitochondrial Fission through the JNK-Drp1 Signaling Pathway
Abstract
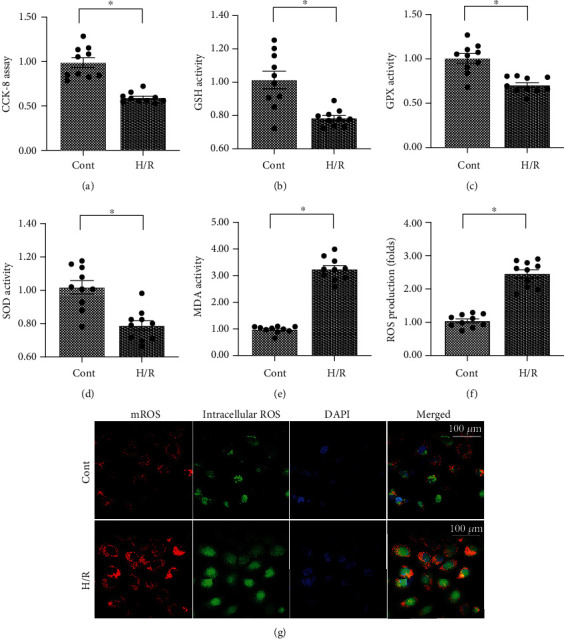
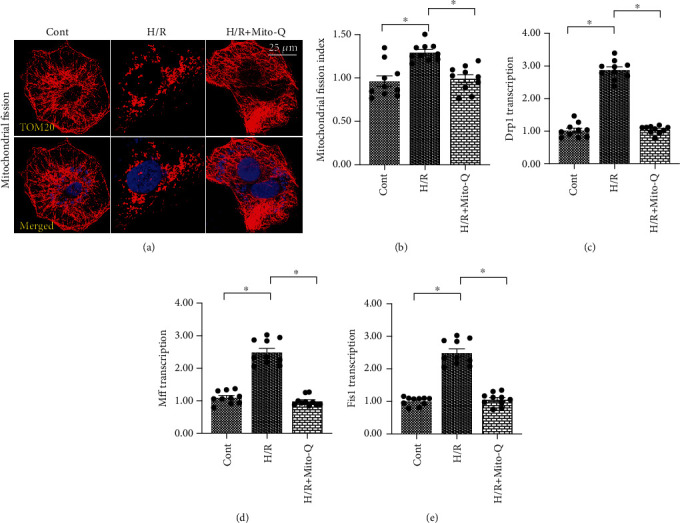
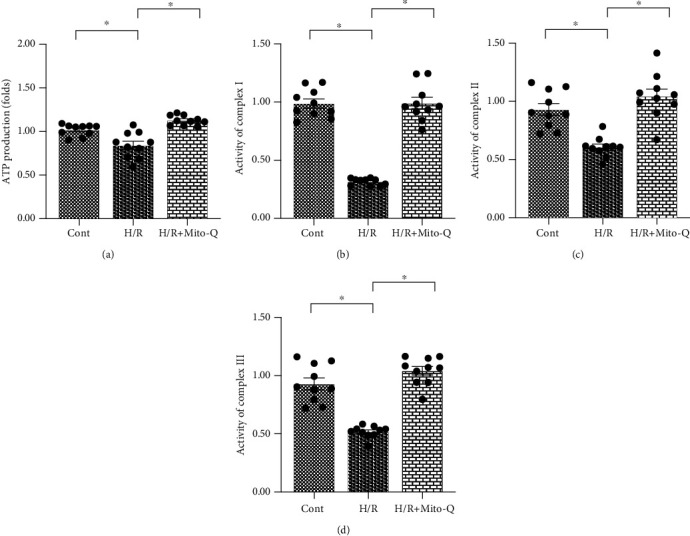
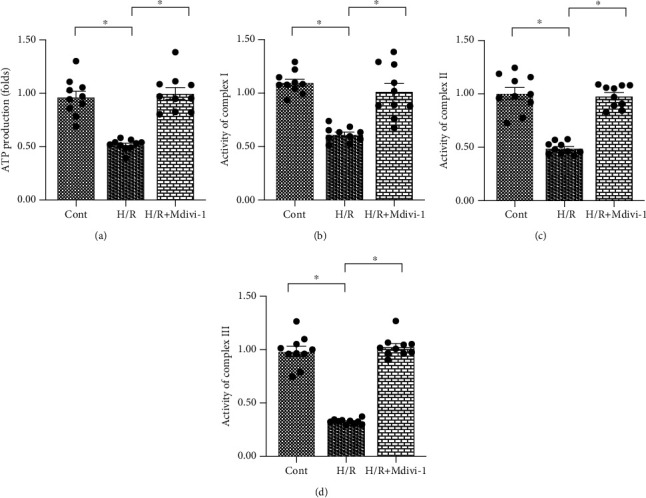
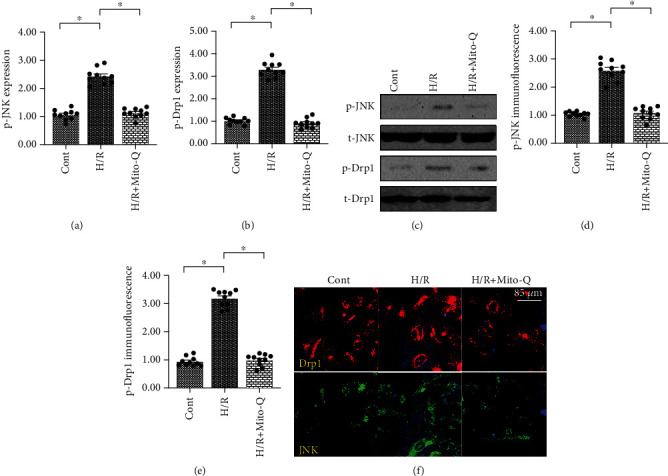
Coronary artery no-reflow is a complex problem in the area of reperfusion therapy, and the molecular mechanisms underlying coronary artery no-reflow injury have not been fully elucidated. In the present study, we explored whether oxidative stress caused damage to coronary endothelial cells by inducing mitochondrial fission and activating the JNK pathway. The hypoxia/reoxygenation (H/R) model was induced in vitro to mimic coronary endothelial no-reflow injury, and mitochondrial fission, mitochondrial function, and endothelial cell viability were analyzed using western blotting, quantitative polymerase chain reaction (qPCR), enzyme-linked immunosorbent assay (ELISA), and immunofluorescence. Our data indicated that reactive oxygen species (ROS) were significantly induced upon H/R injury, and this was followed by decreased endothelial cell viability. Mitochondrial fission was induced and mitochondrial bioenergetics were impaired in cardiac endothelial cells after H/R injury. Neutralization of ROS reduced mitochondrial fission and protected mitochondrial function against H/R injury. Our results also demonstrated that ROS stimulated mitochondrial fission via JNK-mediated Drp1 phosphorylation. These findings indicate that the ROS-JNK-Drp1 signaling pathway may be one of the molecular mechanisms underlying endothelial cell damage during H/R injury. Novel treatments for coronary no-reflow injury may involve targeting mitochondrial fission and the JNK-Drp1 signaling pathway.
Copyright © 2021 Yi Chen et al.
Conflict of interest statement
The authors declare that there is no conflict of interest regarding the publication of this paper.
Figures
Similar articles
-
PTEN-induced kinase 1-induced dynamin-related protein 1 Ser637 phosphorylation reduces mitochondrial fission and protects against intestinal ischemia reperfusion injury.World J Gastroenterol. 2020 Apr 21;26(15):1758-1774. doi: 10.3748/wjg.v26.i15.1758. World J Gastroenterol. 2020. PMID: 32351292 Free PMC article.
-
Mst1 regulates post-infarction cardiac injury through the JNK-Drp1-mitochondrial fission pathway.Cell Mol Biol Lett. 2018 May 8;23:21. doi: 10.1186/s11658-018-0085-1. eCollection 2018. Cell Mol Biol Lett. 2018. PMID: 29760744 Free PMC article.
-
BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways.Angiogenesis. 2018 Aug;21(3):599-615. doi: 10.1007/s10456-018-9611-z. Epub 2018 Apr 6. Angiogenesis. 2018. PMID: 29623489
-
Neuropathic Pain: the Dysfunction of Drp1, Mitochondria, and ROS Homeostasis.Neurotox Res. 2020 Oct;38(3):553-563. doi: 10.1007/s12640-020-00257-2. Epub 2020 Jul 21. Neurotox Res. 2020. PMID: 32696439 Review.
-
Pathological roles of mitochondrial dysfunction in endothelial cells during the cerebral no-reflow phenomenon: A review.Medicine (Baltimore). 2024 Dec 20;103(51):e40951. doi: 10.1097/MD.0000000000040951. Medicine (Baltimore). 2024. PMID: 39705421 Free PMC article. Review.
Cited by
-
Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management (Review).Exp Ther Med. 2022 Jun;23(6):430. doi: 10.3892/etm.2022.11357. Epub 2022 May 6. Exp Ther Med. 2022. PMID: 35607376 Free PMC article. Review.
-
A newly synthesized flavone avoids COMT-catalyzed methylation and mitigates myocardial ischemia/reperfusion injury in H9C2 cells via JNK and P38 pathways.Iran J Basic Med Sci. 2024;27(4):492-499. doi: 10.22038/IJBMS.2023.74358.16149. Iran J Basic Med Sci. 2024. PMID: 38419895 Free PMC article.
-
Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis.Front Physiol. 2022 Dec 20;13:1084604. doi: 10.3389/fphys.2022.1084604. eCollection 2022. Front Physiol. 2022. PMID: 36605901 Free PMC article. Review.
-
Novel Insights and Current Evidence for Mechanisms of Atherosclerosis: Mitochondrial Dynamics as a Potential Therapeutic Target.Front Cell Dev Biol. 2021 Jul 7;9:673839. doi: 10.3389/fcell.2021.673839. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34307357 Free PMC article. Review.
-
The role and mechanisms of microvascular damage in the ischemic myocardium.Cell Mol Life Sci. 2023 Oct 29;80(11):341. doi: 10.1007/s00018-023-04998-z. Cell Mol Life Sci. 2023. PMID: 37898977 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
