

High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings
- PMID: 33614906
- PMCID: PMC7883231
- DOI: 10.1002/smtd.202000792
High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings
Abstract
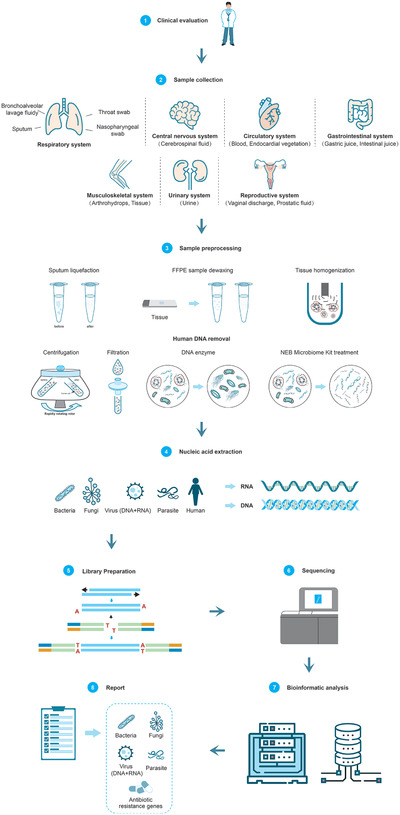
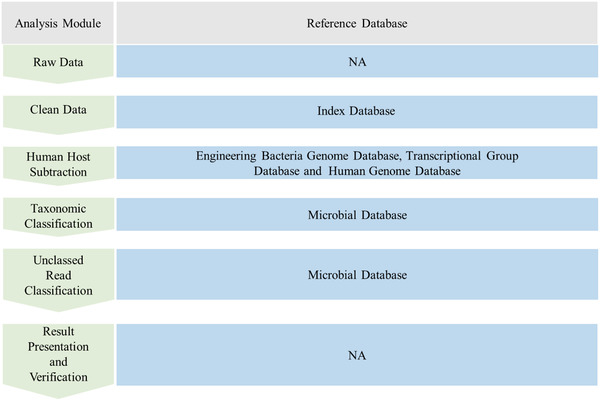
The application of sequencing technology is shifting from research to clinical laboratories owing to rapid technological developments and substantially reduced costs. However, although thousands of microorganisms are known to infect humans, identification of the etiological agents for many diseases remains challenging as only a small proportion of pathogens are identifiable by the current diagnostic methods. These challenges are compounded by the emergence of new pathogens. Hence, metagenomic next-generation sequencing (mNGS), an agnostic, unbiased, and comprehensive method for detection, and taxonomic characterization of microorganisms, has become an attractive strategy. Although many studies, and cases reports, have confirmed the success of mNGS in improving the diagnosis, treatment, and tracking of infectious diseases, several hurdles must still be overcome. It is, therefore, imperative that practitioners and clinicians understand both the benefits and limitations of mNGS when applying it to clinical practice. Interestingly, the emerging third-generation sequencing technologies may partially offset the disadvantages of mNGS. In this review, mainly: a) the history of sequencing technology; b) various NGS technologies, common platforms, and workflows for clinical applications; c) the application of NGS in pathogen identification; d) the global expert consensus on NGS-related methods in clinical applications; and e) challenges associated with diagnostic metagenomics are described.
Keywords: clinical application; infectious disease; metagenomics; next‐generation sequencing.
© 2020 Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
[Results analysis of mNGS applied to infectious diseases].Zhonghua Yu Fang Yi Xue Za Zhi. 2023 Jul 6;57(7):1124-1130. doi: 10.3760/cma.j.cn112150-20220824-00836. Zhonghua Yu Fang Yi Xue Za Zhi. 2023. PMID: 37482746 Review. Chinese.
-
Clinical applications of metagenomics next-generation sequencing in infectious diseases.J Zhejiang Univ Sci B. 2024 May 17;25(6):471-484. doi: 10.1631/jzus.B2300029. J Zhejiang Univ Sci B. 2024. PMID: 38910493 Free PMC article. Review.
-
The clinical application of metagenomic next-generation sequencing for detecting pathogens in bronchoalveolar lavage fluid: case reports and literature review.Expert Rev Mol Diagn. 2022 May;22(5):575-582. doi: 10.1080/14737159.2022.2071607. Epub 2022 May 2. Expert Rev Mol Diagn. 2022. PMID: 35473493 Review.
-
Enhanced Virus Detection and Metagenomic Sequencing in Patients with Meningitis and Encephalitis.mBio. 2021 Aug 31;12(4):e0114321. doi: 10.1128/mBio.01143-21. Epub 2021 Aug 31. mBio. 2021. PMID: 34465023 Free PMC article.
-
Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection.Annu Rev Pathol. 2019 Jan 24;14:319-338. doi: 10.1146/annurev-pathmechdis-012418-012751. Epub 2018 Oct 24. Annu Rev Pathol. 2019. PMID: 30355154 Free PMC article. Review.
Cited by
-
Disseminated Mycobacterium tilburgii infection in a person with AIDS: A case report.Heliyon. 2024 Aug 2;10(15):e35616. doi: 10.1016/j.heliyon.2024.e35616. eCollection 2024 Aug 15. Heliyon. 2024. PMID: 39170256 Free PMC article.
-
Culture-Negative Native Vertebral Osteomyelitis: A Narrative Review of an Underdescribed Condition.J Clin Med. 2024 Sep 28;13(19):5802. doi: 10.3390/jcm13195802. J Clin Med. 2024. PMID: 39407862 Free PMC article. Review.
-
Clinical Application and Influencing Factor Analysis of Metagenomic Next-Generation Sequencing (mNGS) in ICU Patients With Sepsis.Front Cell Infect Microbiol. 2022 Jul 13;12:905132. doi: 10.3389/fcimb.2022.905132. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35909965 Free PMC article.
-
Metagenomic Next-Generation Sequencing for Pulmonary Fungal Infection Diagnosis: Lung Biopsy versus Bronchoalveolar Lavage Fluid.Infect Drug Resist. 2021 Oct 20;14:4333-4359. doi: 10.2147/IDR.S333818. eCollection 2021. Infect Drug Resist. 2021. PMID: 34707378 Free PMC article.
-
Combination of metagenomic next-generation sequencing and conventional tests unraveled pathogen profiles in infected patients undergoing allogeneic hematopoietic stem cell transplantation in Jilin Province of China.Front Cell Infect Microbiol. 2024 Mar 19;14:1378112. doi: 10.3389/fcimb.2024.1378112. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38567023 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
