

Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism
- PMID: 33614908
- PMCID: PMC7873577
- DOI: 10.1016/j.omto.2021.01.001
Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism
Abstract
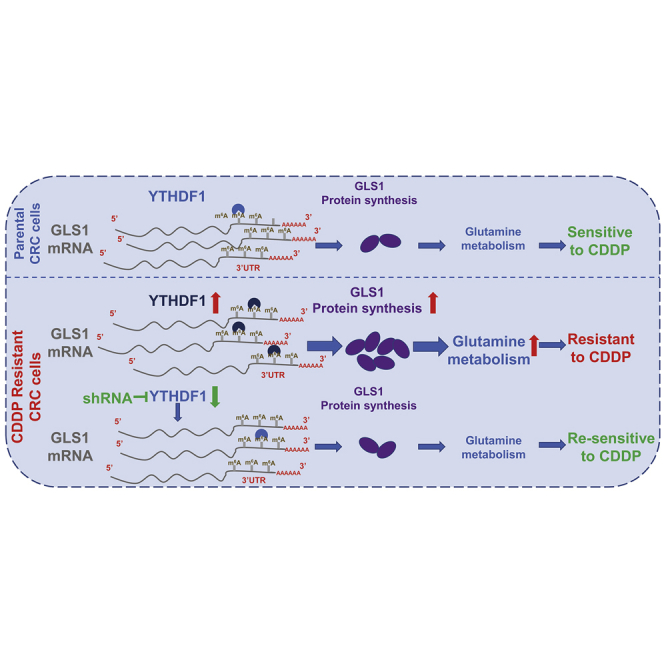
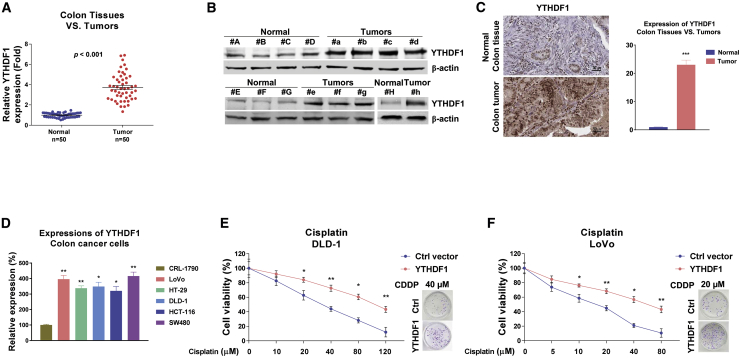
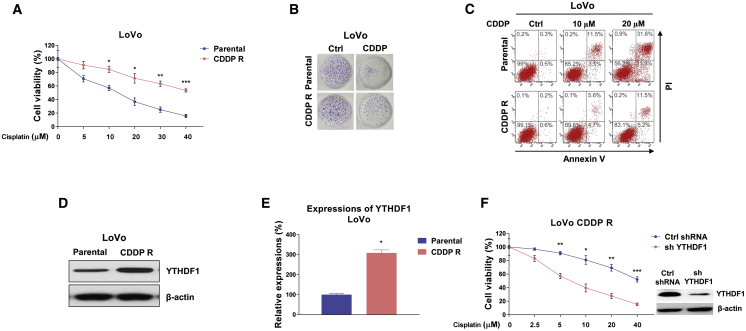
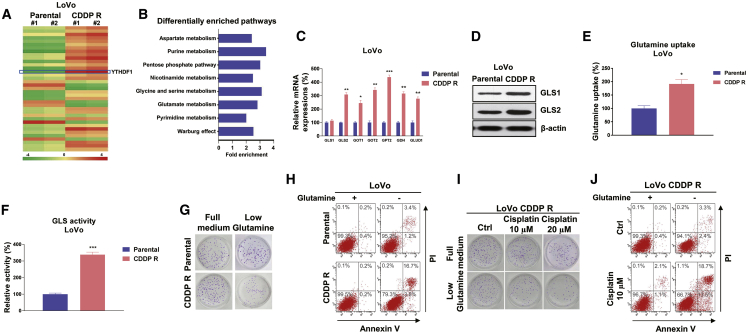
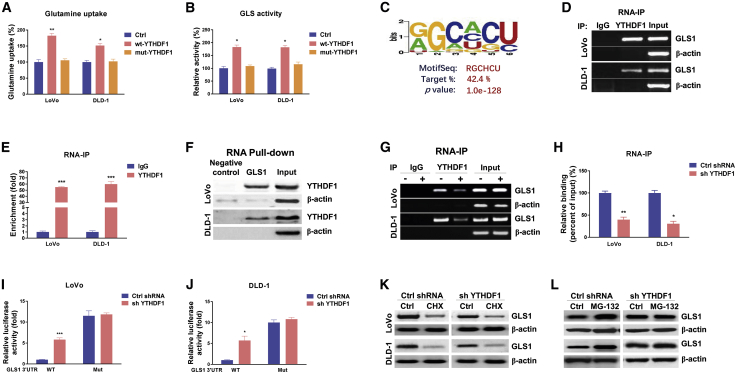
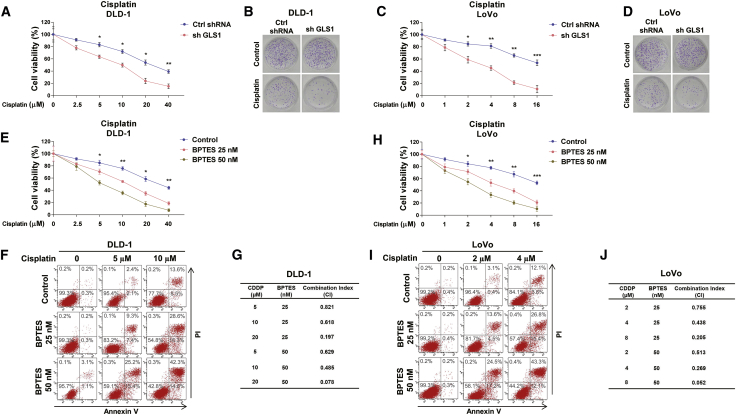
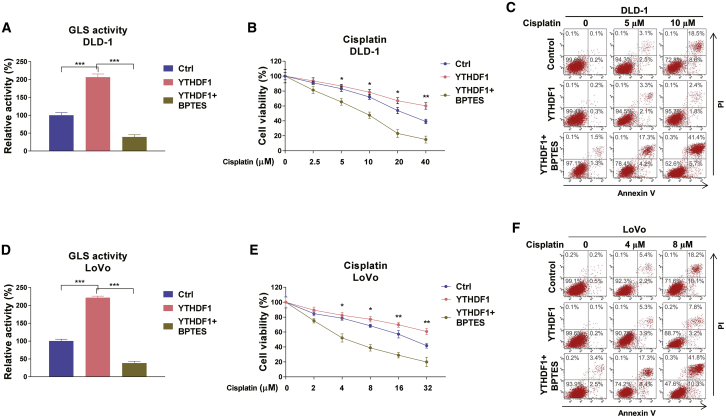
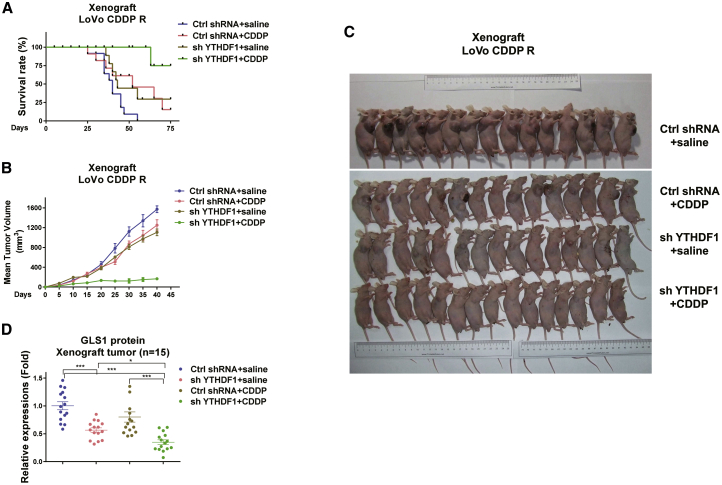
Colorectal cancer (CRC) has a high mortality rate and poor prognosis. Despite chemotherapeutic agents such as cisplatin, which has achieved a better prognosis and survival rate against cancer, drug resistance leads to significant challenges. Accumulating evidence suggests that YTHDF1, the N 6-methyladenosine (m6A) "reader," is an important regulator in tumor progresses. Herein, we report that YTHDF1 was significantly upregulated in human colon tumors and cell lines. Overexpression of YTHDF1 decreased the cisplatin sensitivity of colon cancer cells. From the established cisplatin-resistant CRC cell line (LoVo CDDP R), we detected that YTHDF1 was significantly upregulated in cisplatin-resistant CRC cells. Intriguingly, RNA sequencing (RNA-seq) results revealed that glutamine metabolism enzymes were clearly upregulated in LoVo CDDP R cells. Glutamine uptake, that is, glutaminase (GLS) activity, was upregulated in LoVo CDDP R cells. Furthermore, bioinformatics analysis indicated that the 3' UTR of GLS1 contained a putative binding motif of YTHDF1, and an interaction was further validated by a protein-RNA interaction assay (RNA immunoprecipitation [RIP]). Furthermore, we demonstrated that YTHDF1 promoted protein synthesis of GLS1. Inhibiting GLS1 effectively synergizes with cisplatin to induce colon cancer cell death. Finally, that YTHDF1 mediated cisplatin through the GLS1-glutamine metabolism axis was validated by an in vivo xenograft mouse model. In summary, our study reveals a new mechanism for YTHDF1-promoted cisplatin resistance, contributing to overcoming chemoresistant colon cancers.
© 2021 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
The potential role of m6A reader YTHDF1 as diagnostic biomarker and the signaling pathways in tumorigenesis and metastasis in pan-cancer.Cell Death Discov. 2023 Jan 28;9(1):34. doi: 10.1038/s41420-023-01321-4. Cell Death Discov. 2023. PMID: 36707507 Free PMC article. Review.
-
Curcumin Synergizes with Cisplatin to Inhibit Colon Cancer through Targeting the MicroRNA-137-Glutaminase Axis.Curr Med Sci. 2022 Feb;42(1):108-117. doi: 10.1007/s11596-021-2469-0. Epub 2021 Dec 26. Curr Med Sci. 2022. PMID: 34958454
-
Gegen Qinlian Decoction reverses oxaliplatin resistance in colorectal cancer by inhibiting YTHDF1-regulated m6A modification of GLS1.Phytomedicine. 2024 Oct;133:155906. doi: 10.1016/j.phymed.2024.155906. Epub 2024 Jul 25. Phytomedicine. 2024. PMID: 39089089
-
Targeting PTBP1 blocks glutamine metabolism to improve the cisplatin sensitivity of hepatocarcinoma cells through modulating the mRNA stability of glutaminase.Open Med (Wars). 2023 Sep 12;18(1):20230756. doi: 10.1515/med-2023-0756. eCollection 2023. Open Med (Wars). 2023. PMID: 37724122 Free PMC article.
-
N6-Methyladenosine RNA-Binding Protein YTHDF1 in Gastrointestinal Cancers: Function, Molecular Mechanism and Clinical Implication.Cancers (Basel). 2022 Jul 18;14(14):3489. doi: 10.3390/cancers14143489. Cancers (Basel). 2022. PMID: 35884552 Free PMC article. Review.
Cited by
-
The emerging role of m6A modification of non-coding RNA in gastrointestinal cancers: a comprehensive review.Front Cell Dev Biol. 2023 Oct 30;11:1264552. doi: 10.3389/fcell.2023.1264552. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37965577 Free PMC article. Review.
-
Emerging roles of small extracellular vesicles in metabolic reprogramming and drug resistance in cancers.Cancer Drug Resist. 2024 Sep 27;7:38. doi: 10.20517/cdr.2024.81. eCollection 2024. Cancer Drug Resist. 2024. PMID: 39403606 Free PMC article. Review.
-
Recent Advances in RNA m6A Modification in Solid Tumors and Tumor Immunity.Cancer Treat Res. 2023;190:95-142. doi: 10.1007/978-3-031-45654-1_4. Cancer Treat Res. 2023. PMID: 38113000
-
The potential role of m6A reader YTHDF1 as diagnostic biomarker and the signaling pathways in tumorigenesis and metastasis in pan-cancer.Cell Death Discov. 2023 Jan 28;9(1):34. doi: 10.1038/s41420-023-01321-4. Cell Death Discov. 2023. PMID: 36707507 Free PMC article. Review.
-
A synthesized olean-28,13β-lactam targets YTHDF1-GLS1 axis to induce ROS-dependent metabolic crisis and cell death in pancreatic adenocarcinoma.Cancer Cell Int. 2022 Apr 2;22(1):143. doi: 10.1186/s12935-022-02562-6. Cancer Cell Int. 2022. PMID: 35366902 Free PMC article.
References
-
- Dekker E., Tanis P.J., Vleugels J.L.A., Kasi P.M., Wallace M.B. Colorectal cancer. Lancet. 2019;394:1467–1480. - PubMed
-
- Dienstmann R., Salazar R., Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J. Clin. Oncol. 2015;33:1787–1796. - PubMed
-
- Meyers B.M., Cosby R., Quereshy F., Jonker D. Adjuvant chemotherapy for stage II and III colon cancer following complete resection: a Cancer Care Ontario systematic review. Clin. Oncol. (R. Coll. Radiol.) 2017;29:459–465. - PubMed
-
- Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
