

AKAP79/150 coordinates leptin-induced PKA signaling to regulate KATP channel trafficking in pancreatic β-cells
- PMID: 33617875
- PMCID: PMC8010710
- DOI: 10.1016/j.jbc.2021.100442
AKAP79/150 coordinates leptin-induced PKA signaling to regulate KATP channel trafficking in pancreatic β-cells
Abstract
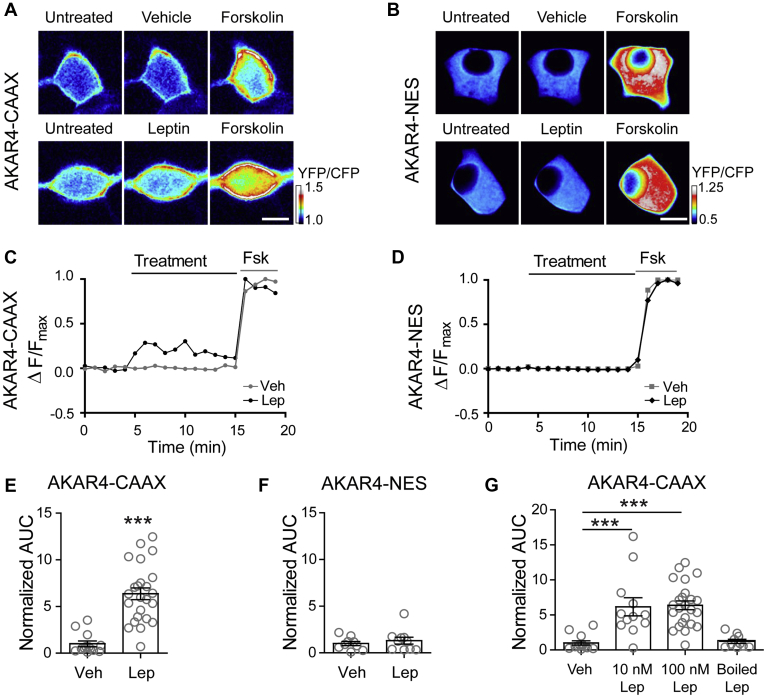
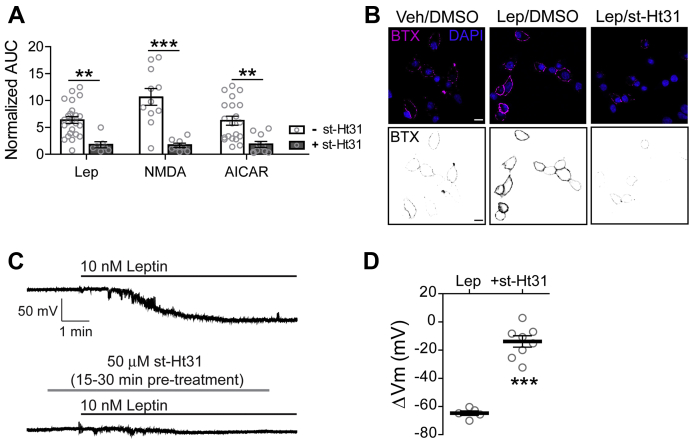
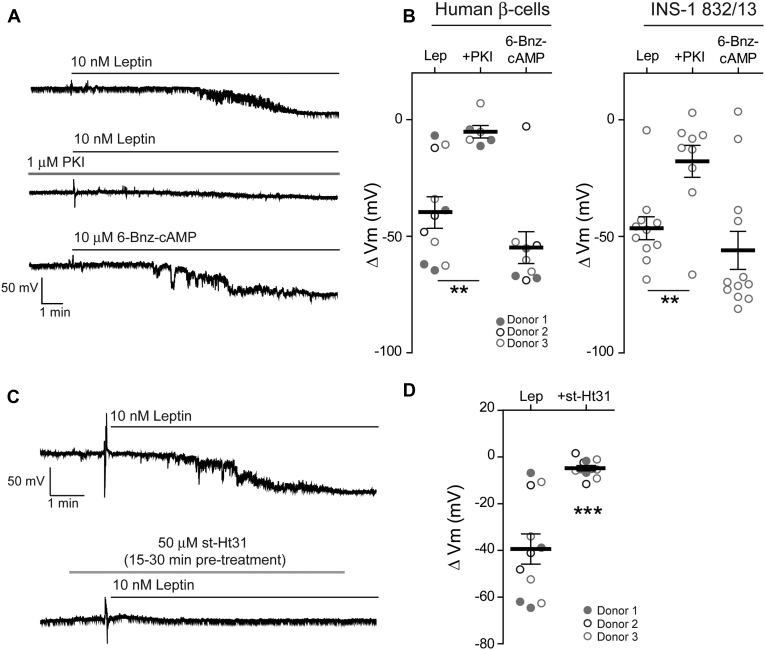
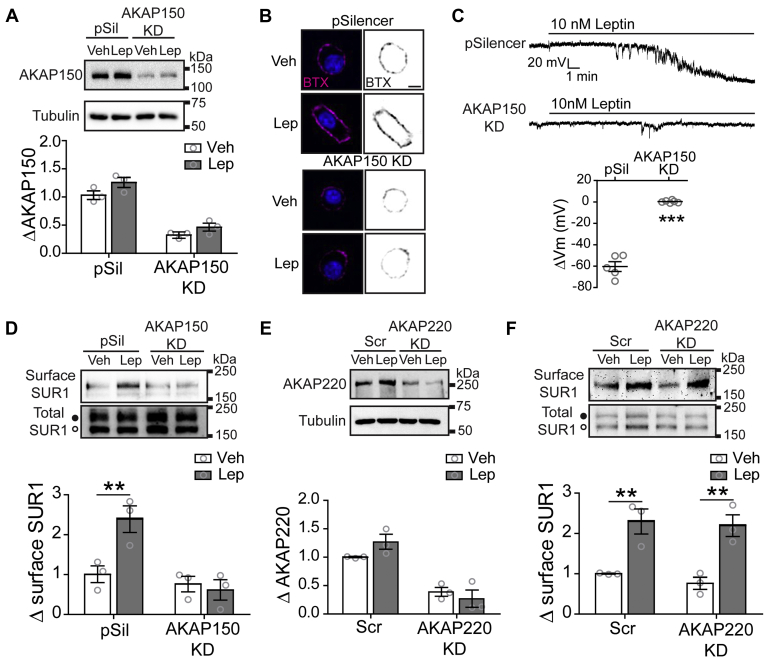
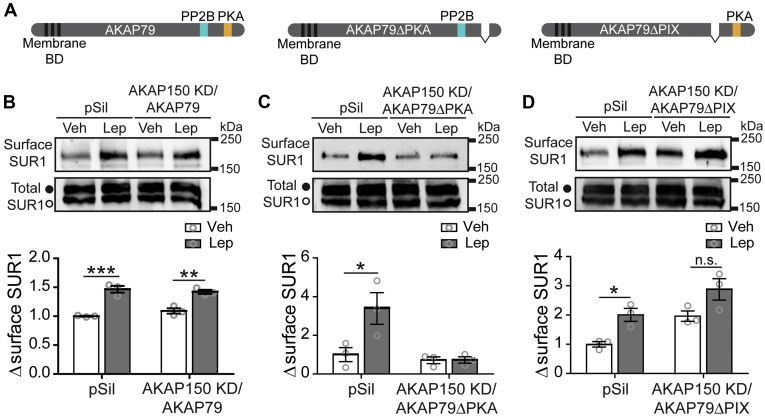
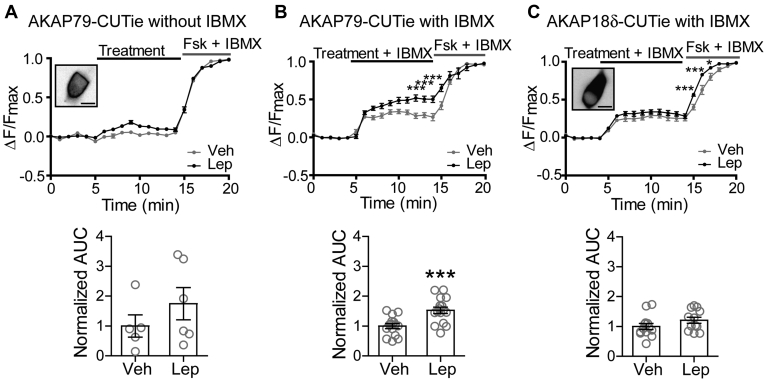
The adipocyte hormone leptin regulates glucose homeostasis both centrally and peripherally. A key peripheral target is the pancreatic β-cell, which secretes insulin upon glucose stimulation. Leptin is known to suppress glucose-stimulated insulin secretion by promoting trafficking of KATP channels to the β-cell surface, which increases K+ conductance and causes β-cell hyperpolarization. We have previously shown that leptin-induced KATP channel trafficking requires protein kinase A (PKA)-dependent actin remodeling. However, whether PKA is a downstream effector of leptin signaling or PKA plays a permissive role is unknown. Using FRET-based reporters of PKA activity, we show that leptin increases PKA activity at the cell membrane and that this effect is dependent on N-methyl-D-aspartate receptors, CaMKKβ, and AMPK, which are known to be involved in the leptin signaling pathway. Genetic knockdown and rescue experiments reveal that the increased PKA activity upon leptin stimulation requires the membrane-targeted PKA-anchoring protein AKAP79/150, indicating that PKA activated by leptin is anchored to AKAP79/150. Interestingly, disrupting protein phosphatase 2B (PP2B) anchoring to AKAP79/150, known to elevate basal PKA signaling, leads to increased surface KATP channels even in the absence of leptin stimulation. Our findings uncover a novel role of AKAP79/150 in coordinating leptin and PKA signaling to regulate KATP channel trafficking in β-cells, hence insulin secretion. The study further advances our knowledge of the downstream signaling events that may be targeted to restore insulin secretion regulation in β-cells defective in leptin signaling, such as those from obese individuals with type 2 diabetes.
Keywords: A-kinase anchoring protein (AKAP); ATP-sensitive potassium channel; cyclic AMP (cAMP); fluorescence resonance energy transfer (FRET); leptin; protein kinase A (PKA).
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures


References
-
- Emilsson V., Liu Y.L., Cawthorne M. a, Morton N.M., Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. - PubMed
-
- Ookuma K., Ookuma M., York D.A. Effects of leptin on insulin secretion from isolated rat pancreatic islets. Diabetes. 1998;47:219–223. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
