

Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase
- PMID: 33618748
- PMCID: PMC7898412
- DOI: 10.1186/s13059-021-02283-5
Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase
Abstract
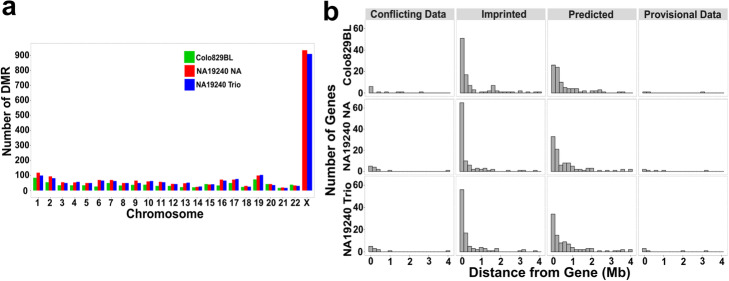
The ability of nanopore sequencing to simultaneously detect modified nucleotides while producing long reads makes it ideal for detecting and phasing allele-specific methylation. However, there is currently no complete software for detecting SNPs, phasing haplotypes, and mapping methylation to these from nanopore sequence data. Here, we present NanoMethPhase, a software tool to phase 5-methylcytosine from nanopore sequencing. We also present SNVoter, which can post-process nanopore SNV calls to improve accuracy in low coverage regions. Together, these tools can accurately detect allele-specific methylation genome-wide using nanopore sequence data with low coverage of about ten-fold redundancy.
Keywords: Allele-specific methylation; NanoMethPhase; Nanopore sequencing; Phasing.
Conflict of interest statement
We declare that there is no conflict of interest associated with this publication.
Figures
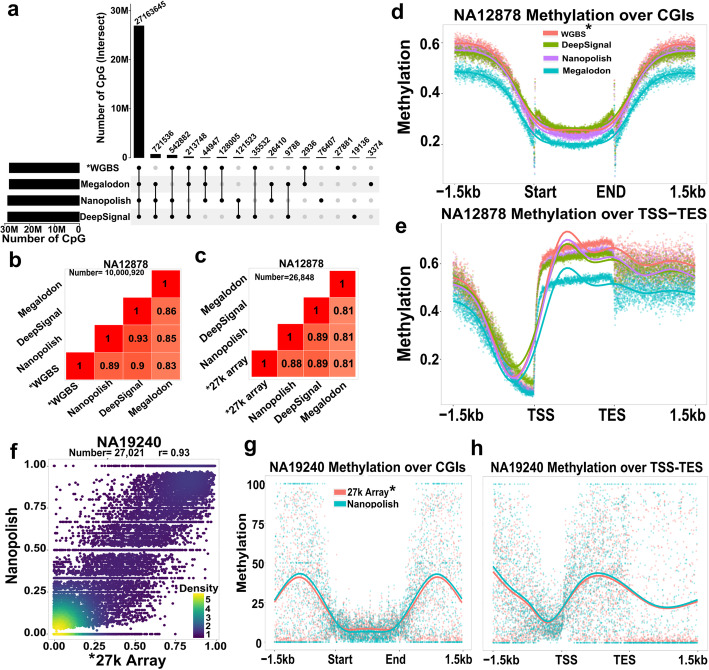
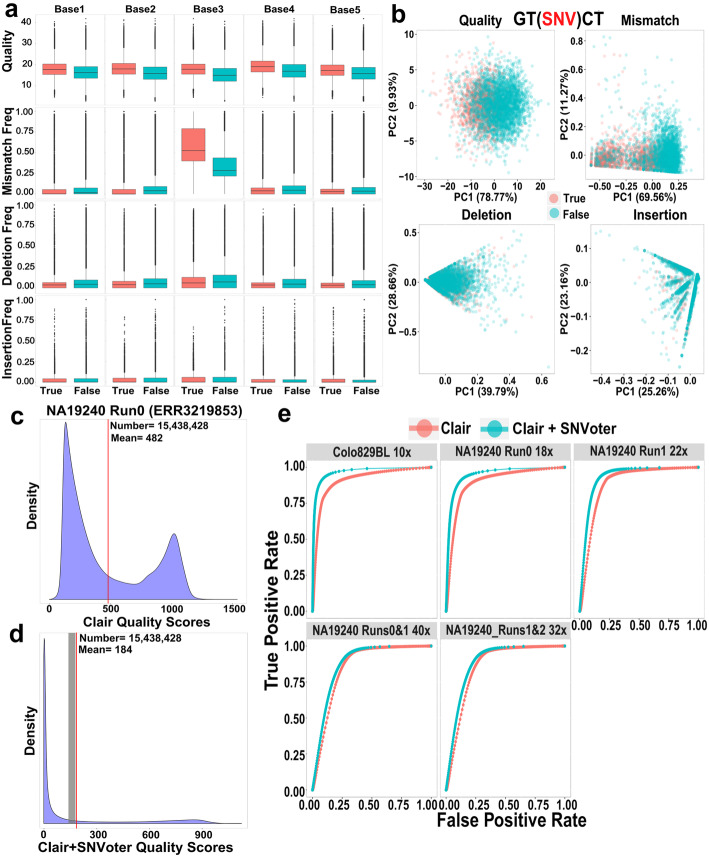
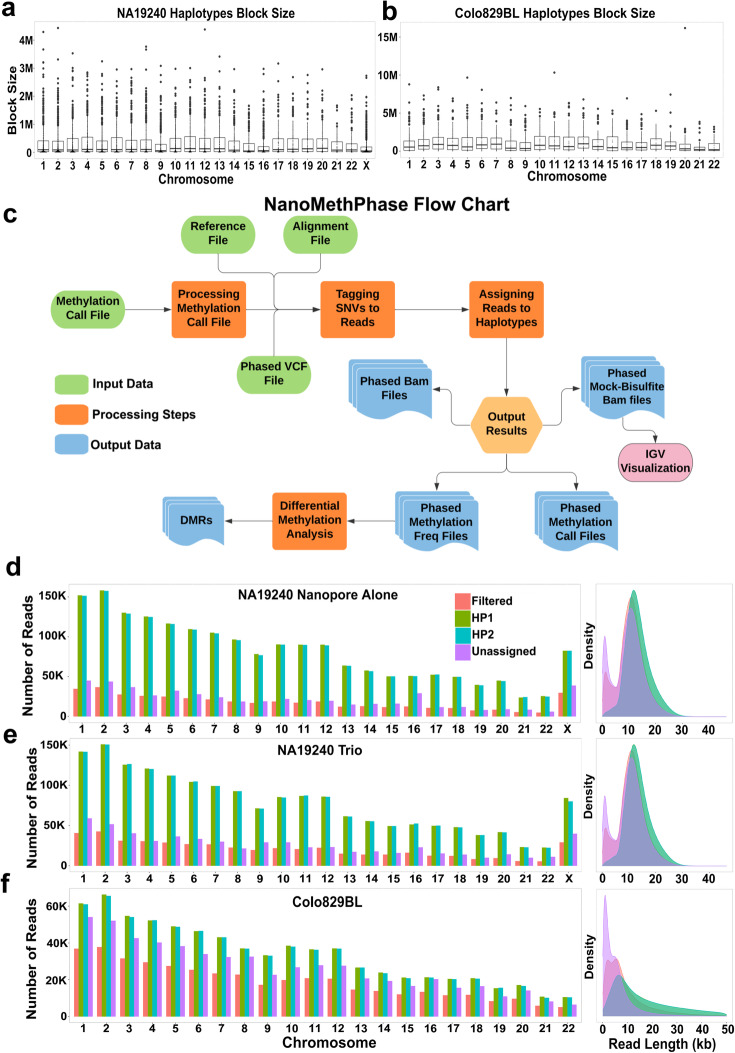
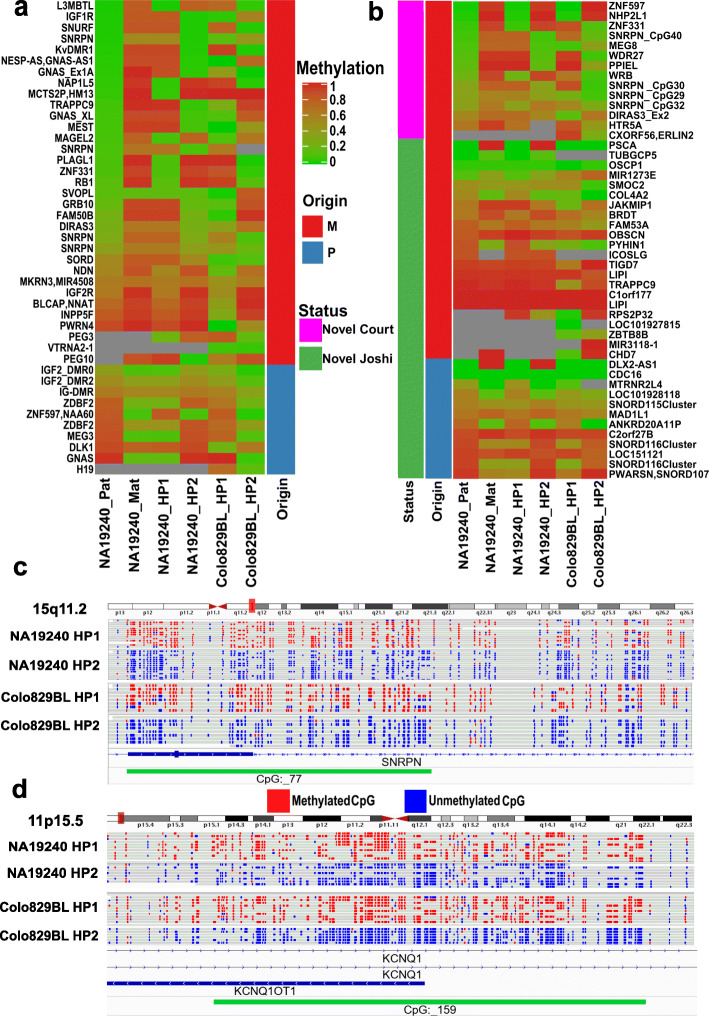
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
