

Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients
- PMID: 33618765
- PMCID: PMC7898256
- DOI: 10.1186/s13073-021-00847-5
Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients
Abstract
Background: Since early February 2021, the causative agent of COVID-19, SARS-CoV-2, has infected over 104 million people with more than 2 million deaths according to official reports. The key to understanding the biology and virus-host interactions of SARS-CoV-2 requires the knowledge of mutation and evolution of this virus at both inter- and intra-host levels. However, despite quite a few polymorphic sites identified among SARS-CoV-2 populations, intra-host variant spectra and their evolutionary dynamics remain mostly unknown.
Methods: Using high-throughput sequencing of metatranscriptomic and hybrid captured libraries, we characterized consensus genomes and intra-host single nucleotide variations (iSNVs) of serial samples collected from eight patients with COVID-19. The distribution of iSNVs along the SARS-CoV-2 genome was analyzed and co-occurring iSNVs among COVID-19 patients were identified. We also compared the evolutionary dynamics of SARS-CoV-2 population in the respiratory tract (RT) and gastrointestinal tract (GIT).
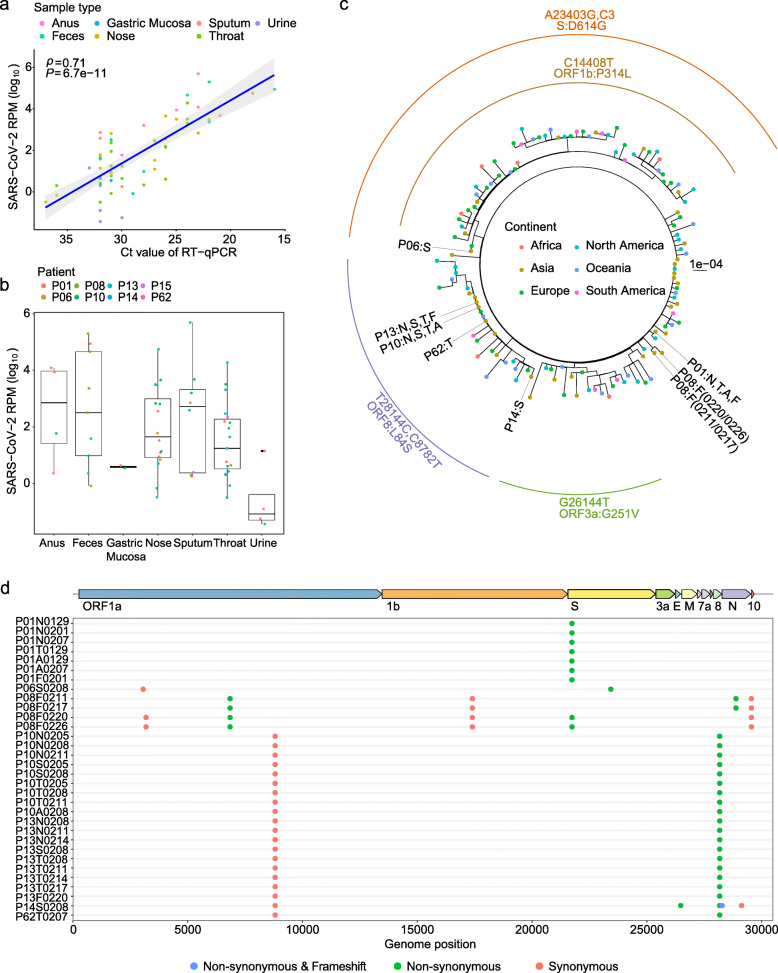
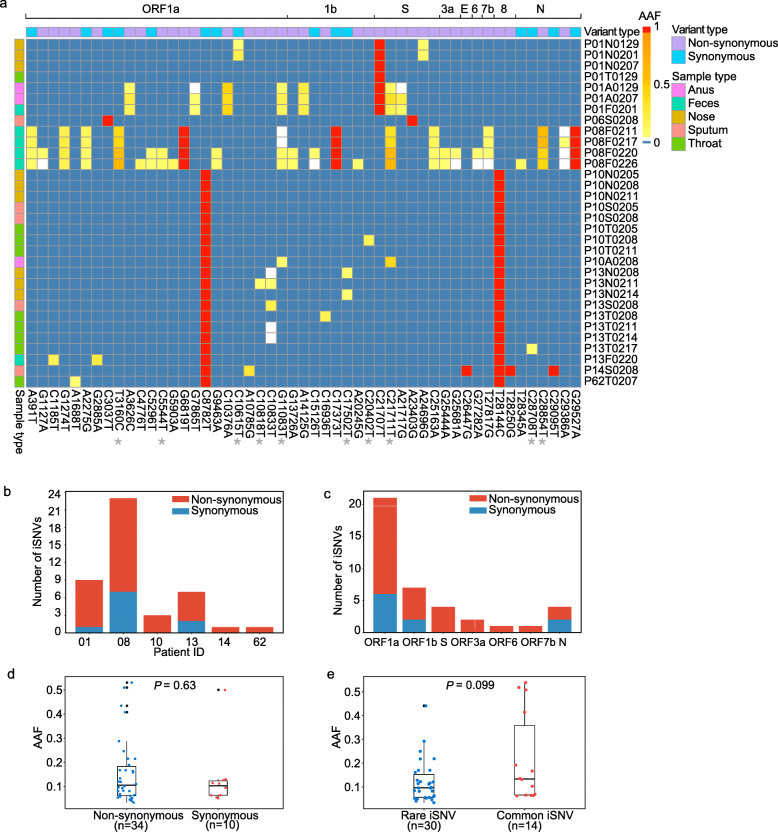
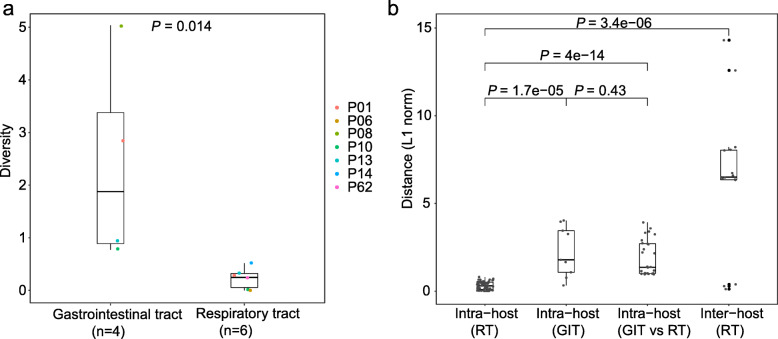
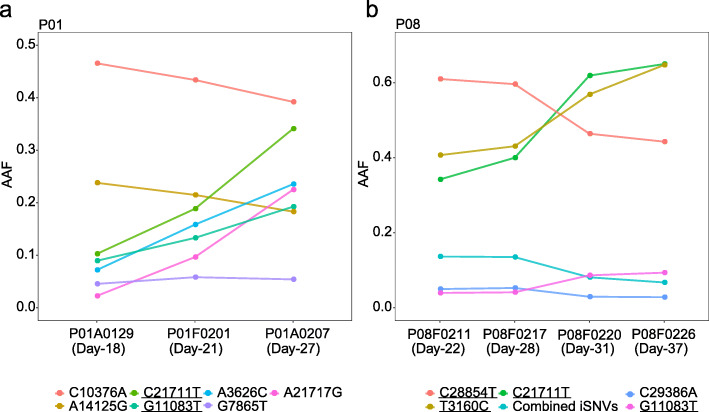
Results: The 32 consensus genomes revealed the co-existence of different genotypes within the same patient. We further identified 40 intra-host single nucleotide variants (iSNVs). Most (30/40) iSNVs presented in a single patient, while ten iSNVs were found in at least two patients or identical to consensus variants. Comparing allele frequencies of the iSNVs revealed a clear genetic differentiation between intra-host populations from the respiratory tract (RT) and gastrointestinal tract (GIT), mostly driven by bottleneck events during intra-host migrations. Compared to RT populations, the GIT populations showed a better maintenance and rapid development of viral genetic diversity following the suspected intra-host bottlenecks.
Conclusions: Our findings here illustrate the intra-host bottlenecks and evolutionary dynamics of SARS-CoV-2 in different anatomic sites and may provide new insights to understand the virus-host interactions of coronaviruses and other RNA viruses.
Keywords: COVID-19; Dynamics; Intra-host; SARS-CoV-2; Variation.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- 2018YFC1200100, 2018ZX10301403, 2018YFC1311900/The National Key Research and Development Program of China
- 2020YFC0841400/the emergency grants for prevention and control of SARS-CoV-2 of Ministry of Science and Technology
- 2020B1111320003, 2020B111108001, 2018B020207013, 2020B111112003/the emergency grants for prevention and control of SARS-CoV-2 of Guangdong province
- 2020A1515010911/the Guangdong Province Basic and Applied Basic Research Fund
- 2019B030316028/Guangdong Science and Technology Foundation
- 2017B030301011/Guangdong Provincial Key Laboratory of Genome Read and Write
- [2017] No.159/Guangzhou Medical University High-level University Innovation Team Training Program
- 81702047, 81772191, 91842106 and 8181101118/National Natural Science Foundation of China
- SKLRD-QN-201715, SKLRD-QN-201912 and SKLRD-Z-202007/State Key Laboratory of Respiratory Disease
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
