

CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores
- PMID: 33618777
- PMCID: PMC7901104
- DOI: 10.1186/s13073-021-00835-9
CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores
Abstract
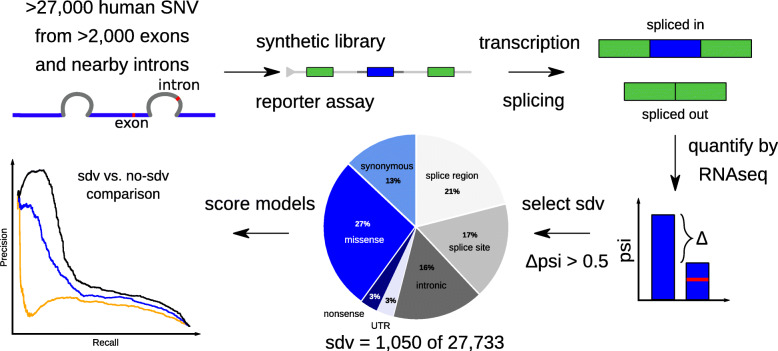
Background: Splicing of genomic exons into mRNAs is a critical prerequisite for the accurate synthesis of human proteins. Genetic variants impacting splicing underlie a substantial proportion of genetic disease, but are challenging to identify beyond those occurring at donor and acceptor dinucleotides. To address this, various methods aim to predict variant effects on splicing. Recently, deep neural networks (DNNs) have been shown to achieve better results in predicting splice variants than other strategies.
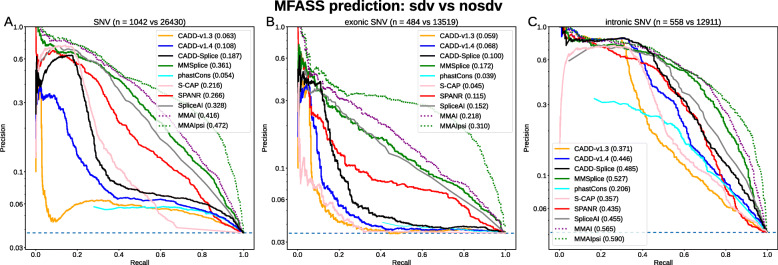
Methods: It has been unclear how best to integrate such process-specific scores into genome-wide variant effect predictors. Here, we use a recently published experimental data set to compare several machine learning methods that score variant effects on splicing. We integrate the best of those approaches into general variant effect prediction models and observe the effect on classification of known pathogenic variants.
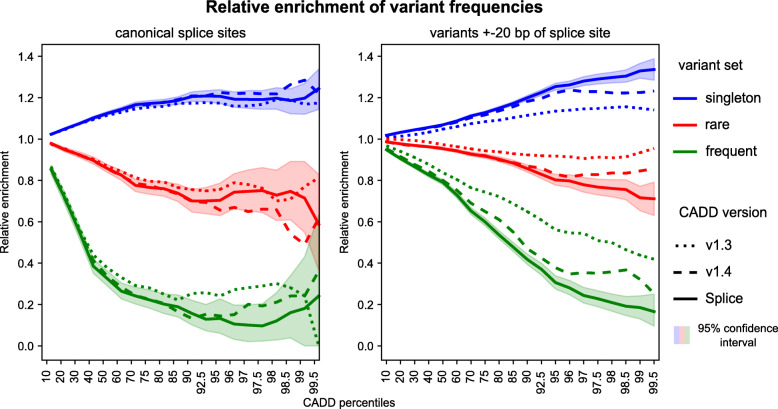
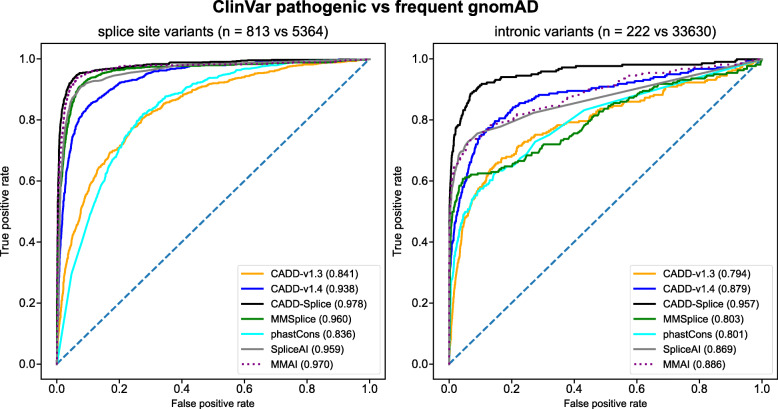
Results: We integrate two specialized splicing scores into CADD (Combined Annotation Dependent Depletion; cadd.gs.washington.edu ), a widely used tool for genome-wide variant effect prediction that we previously developed to weight and integrate diverse collections of genomic annotations. With this new model, CADD-Splice, we show that inclusion of splicing DNN effect scores substantially improves predictions across multiple variant categories, without compromising overall performance.
Conclusions: While splice effect scores show superior performance on splice variants, specialized predictors cannot compete with other variant scores in general variant interpretation, as the latter account for nonsense and missense effects that do not alter splicing. Although only shown here for splice scores, we believe that the applied approach will generalize to other specific molecular processes, providing a path for the further improvement of genome-wide variant effect prediction.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
