

Cell Type-specific Adaptive Signaling Responses to KRASG12C Inhibition
- PMID: 33619172
- PMCID: PMC9940280
- DOI: 10.1158/1078-0432.CCR-20-3872
Cell Type-specific Adaptive Signaling Responses to KRASG12C Inhibition
Abstract
Purpose: Covalent inhibitors of KRASG12C specifically target tumors driven by this form of mutant KRAS, yet early studies show that bypass signaling drives adaptive resistance. Although several combination strategies have been shown to improve efficacy of KRASG12C inhibitors (KRASi), underlying mechanisms and predictive strategies for patient enrichment are less clear.
Experimental design: We performed mass spectrometry-based phosphoproteomics analysis in KRASG12C cell lines after short-term treatment with ARS-1620. To understand signaling diversity and cell type-specific markers, we compared proteome and phosphoproteomes of KRASG12C cells. Gene expression patterns of KRASG12C cell lines and lung tumor tissues were examined.
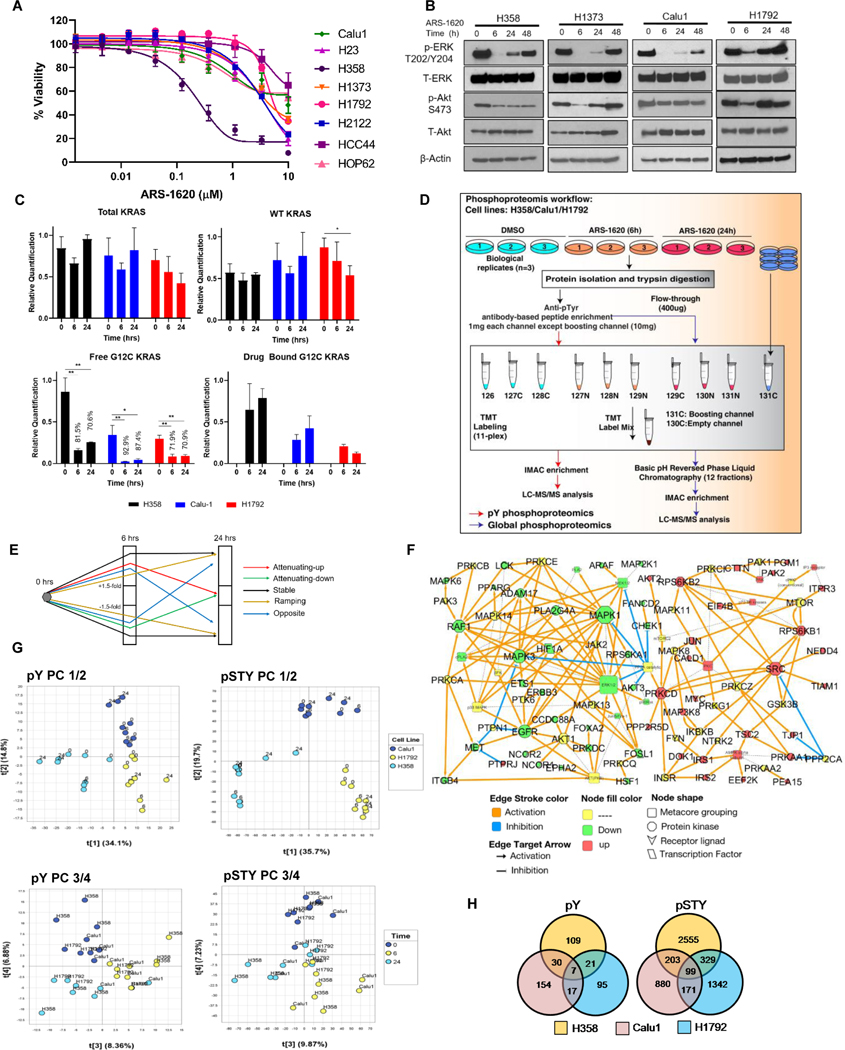
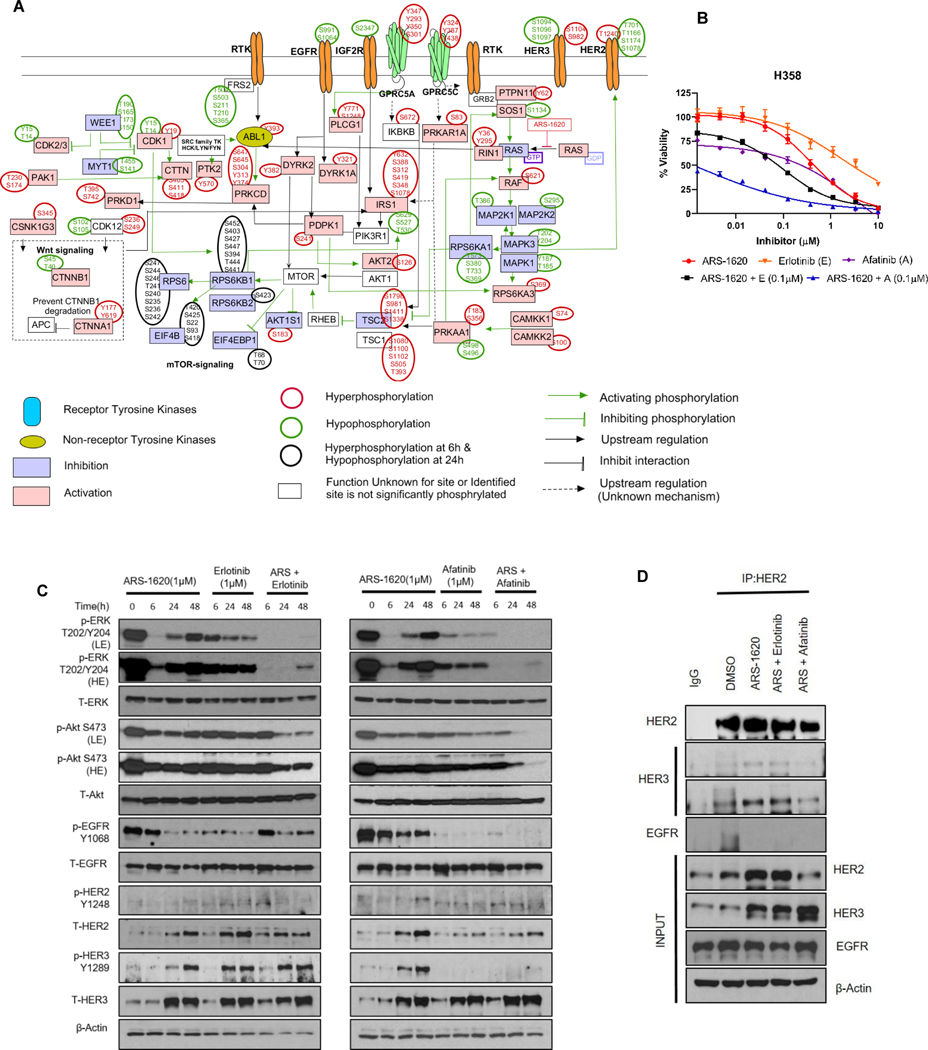
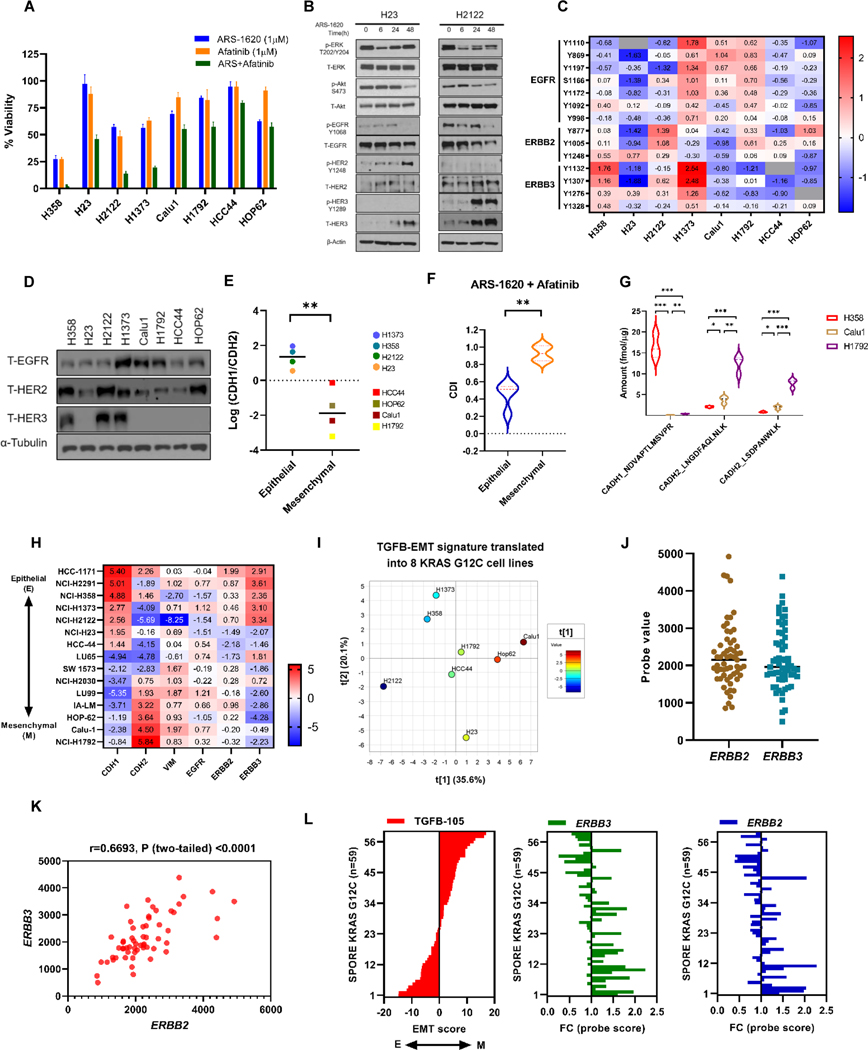
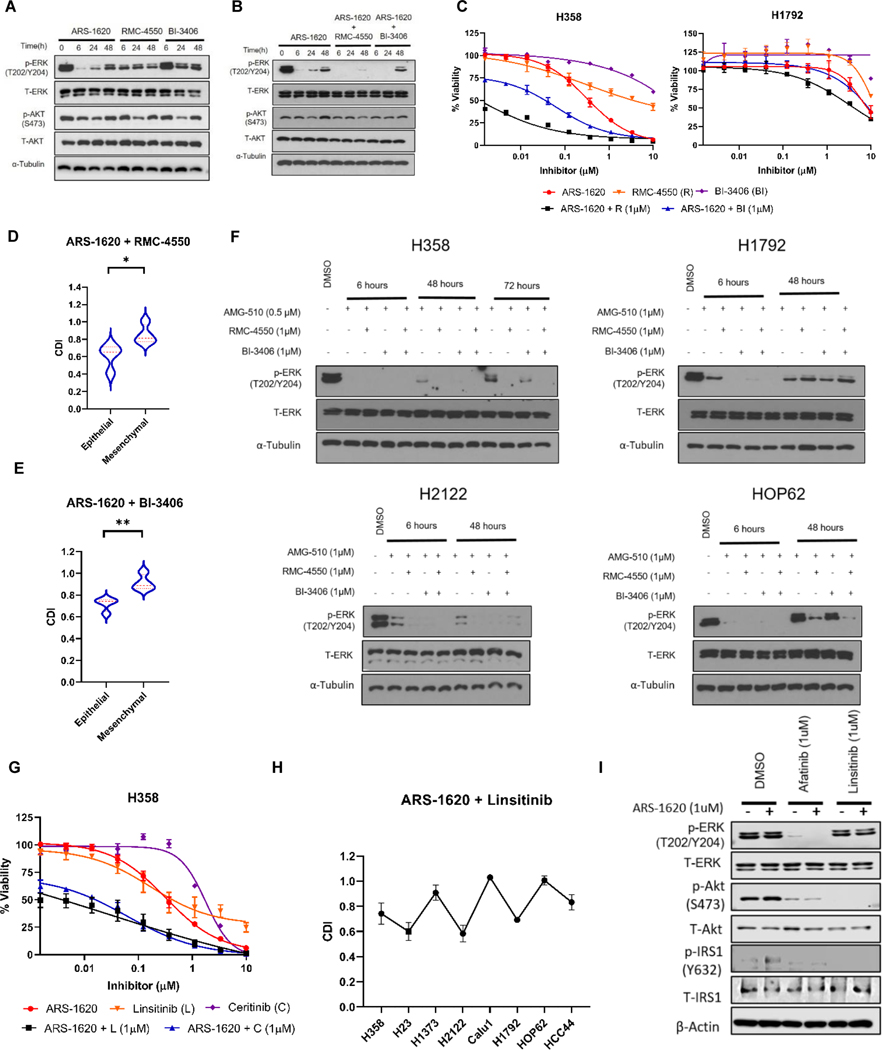
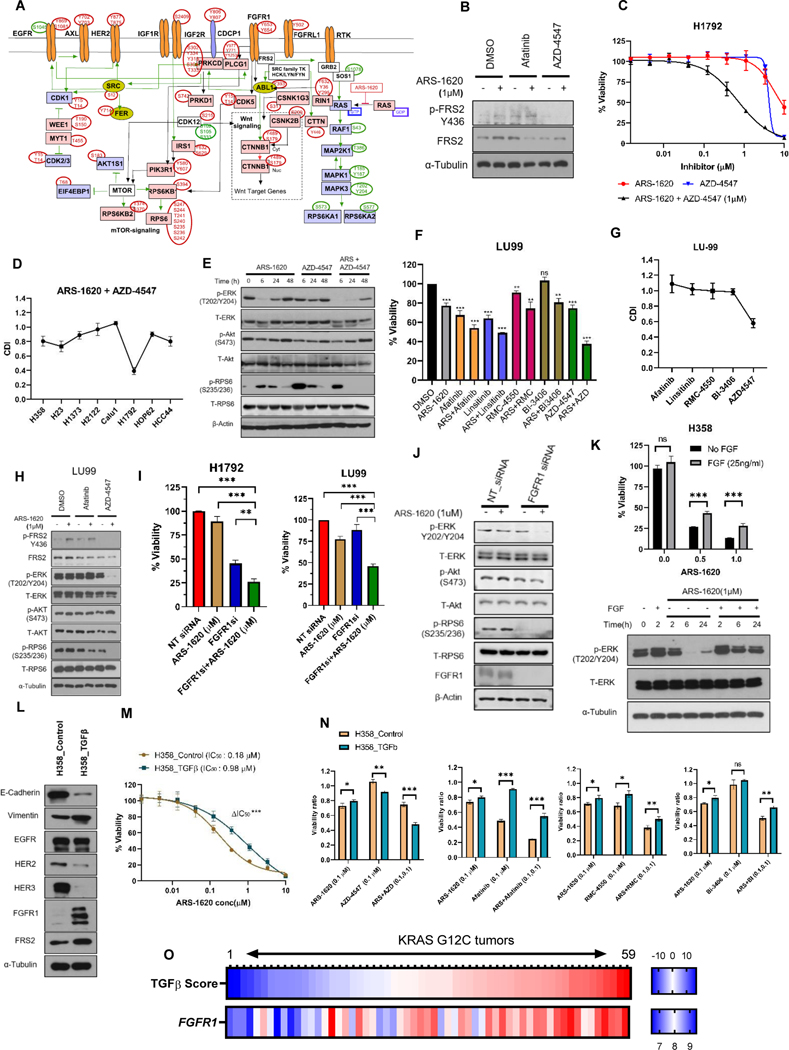
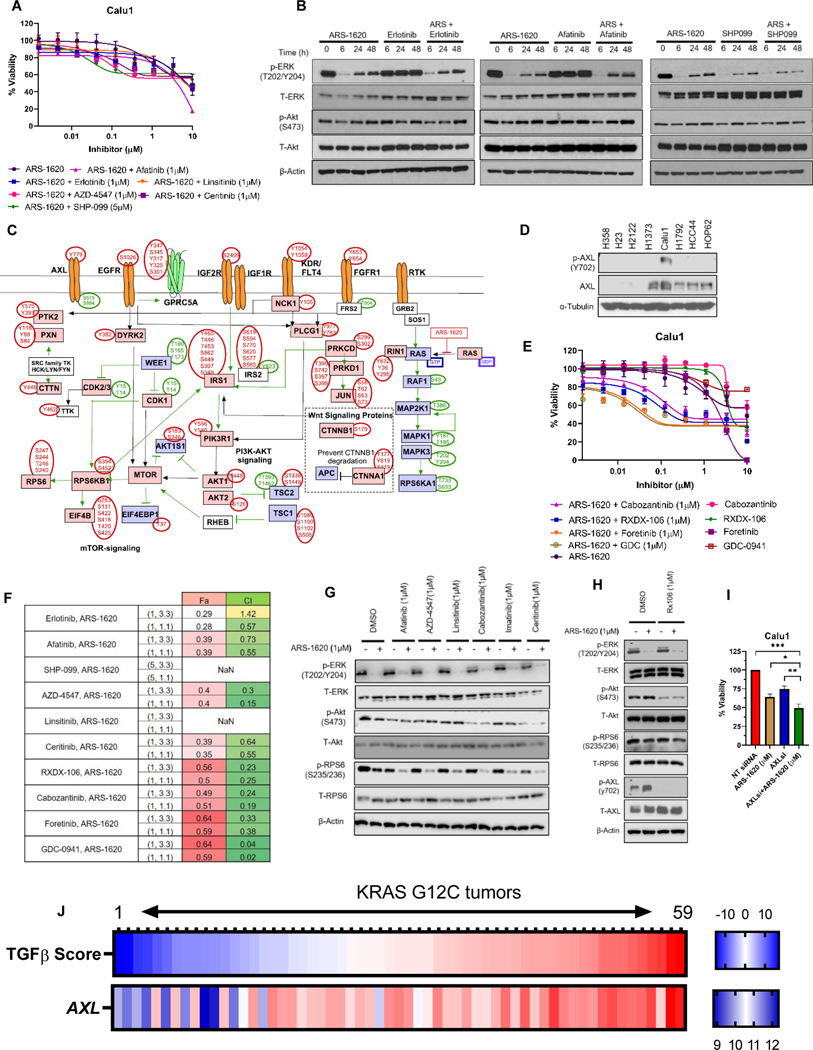
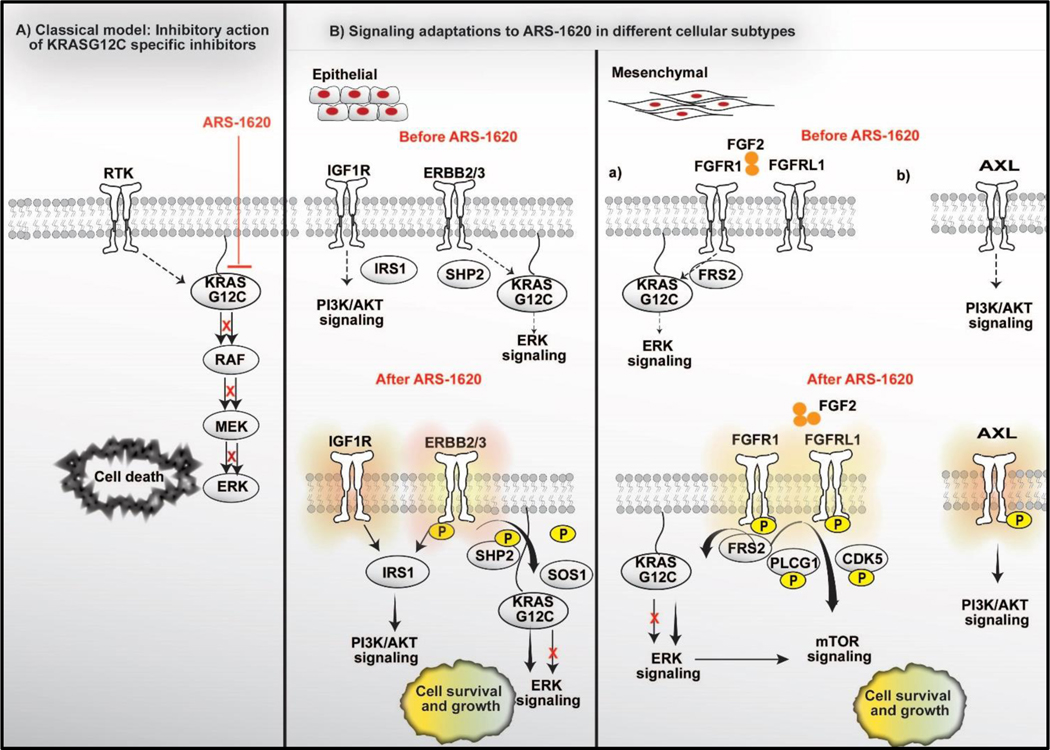
Results: Our analysis suggests cell type-specific perturbation to ERBB2/3 signaling compensates for repressed ERK and AKT signaling following ARS-1620 treatment in epithelial cell type, and this subtype was also more responsive to coinhibition of SHP2 and SOS1. Conversely, both high basal and feedback activation of FGFR or AXL signaling were identified in mesenchymal cells. Inhibition of FGFR signaling suppressed feedback activation of ERK and mTOR, while AXL inhibition suppressed PI3K pathway. In both cell lines and human lung cancer tissues with KRASG12C, we observed high basal ERBB2/3 associated with epithelial gene signatures, while higher basal FGFR1 and AXL were observed in cells/tumors with mesenchymal gene signatures.
Conclusions: Our phosphoproteomic study identified cell type-adaptive responses to KRASi. Markers and targets associated with ERBB2/3 signaling in epithelial subtype and with FGFR1/AXL signaling in mesenchymal subtype should be considered in patient enrichment schemes with KRASi.
©2021 American Association for Cancer Research.
Figures
References
-
- Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res 2015;21(4):730–8 doi 10.1158/1078-0432.CCR-14-1814. - DOI - PubMed
-
- Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 2016;6(3):316–29 doi 10.1158/2159-8290.CD-15-1105. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
