

Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2
- PMID: 33619331
- PMCID: PMC7900180
- DOI: 10.1038/s41598-021-83761-5
Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2
Abstract
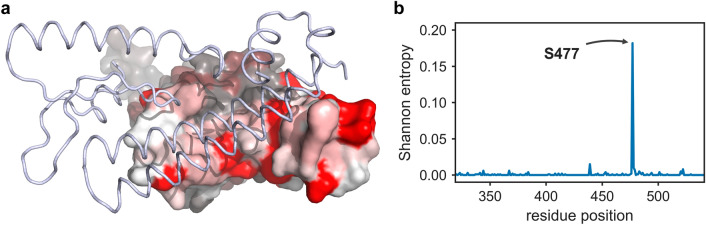
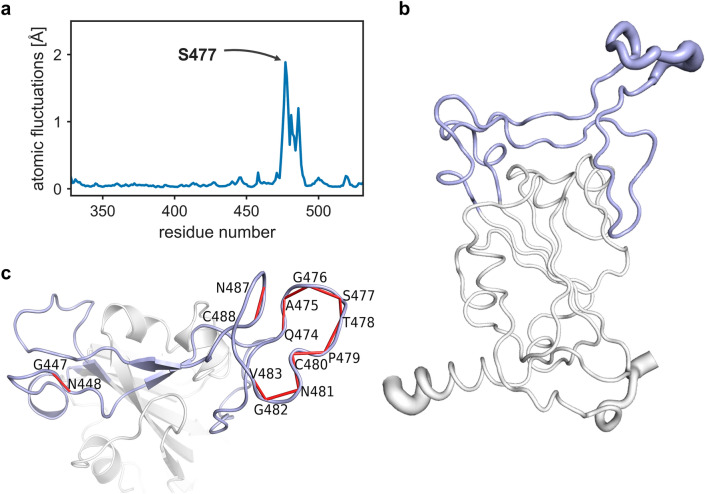
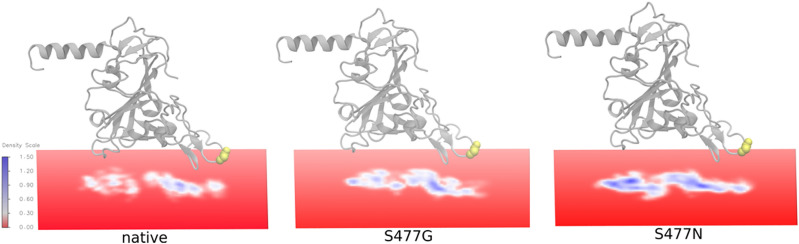
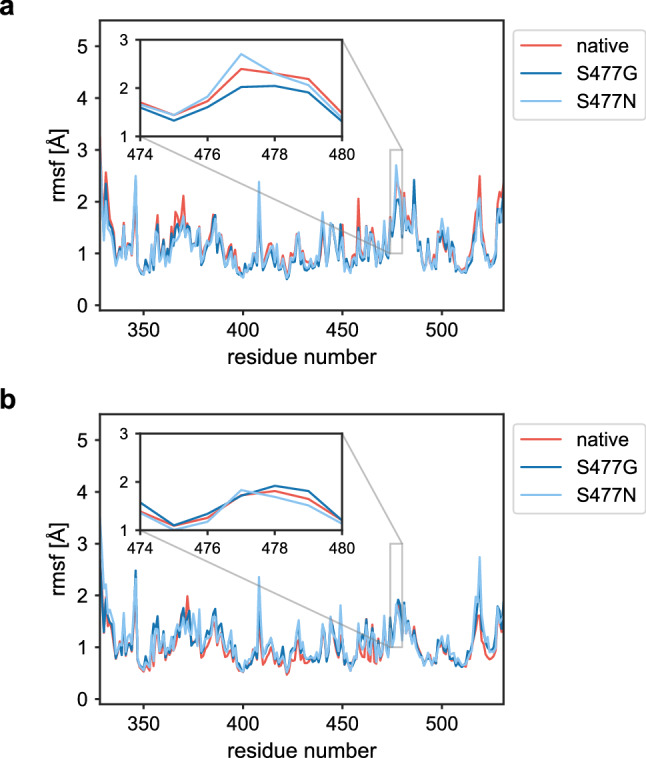
Since the worldwide outbreak of the infectious disease COVID-19, several studies have been published to understand the structural mechanism of the novel coronavirus SARS-CoV-2. During the infection process, the SARS-CoV-2 spike (S) protein plays a crucial role in the receptor recognition and cell membrane fusion process by interacting with the human angiotensin-converting enzyme 2 (hACE2) receptor. However, new variants of these spike proteins emerge as the virus passes through the disease reservoir. This poses a major challenge for designing a potent antigen for an effective immune response against the spike protein. Through a normal mode analysis (NMA) we identified the highly flexible region in the receptor binding domain (RBD) of SARS-CoV-2, starting from residue 475 up to residue 485. Structurally, the position S477 shows the highest flexibility among them. At the same time, S477 is hitherto the most frequently exchanged amino acid residue in the RBDs of SARS-CoV-2 mutants. Therefore, using MD simulations, we have investigated the role of S477 and its two frequent mutations (S477G and S477N) at the RBD during the binding to hACE2. We found that the amino acid exchanges S477G and S477N strengthen the binding of the SARS-COV-2 spike with the hACE2 receptor.
Conflict of interest statement
The authors declare no competing interests.
Figures
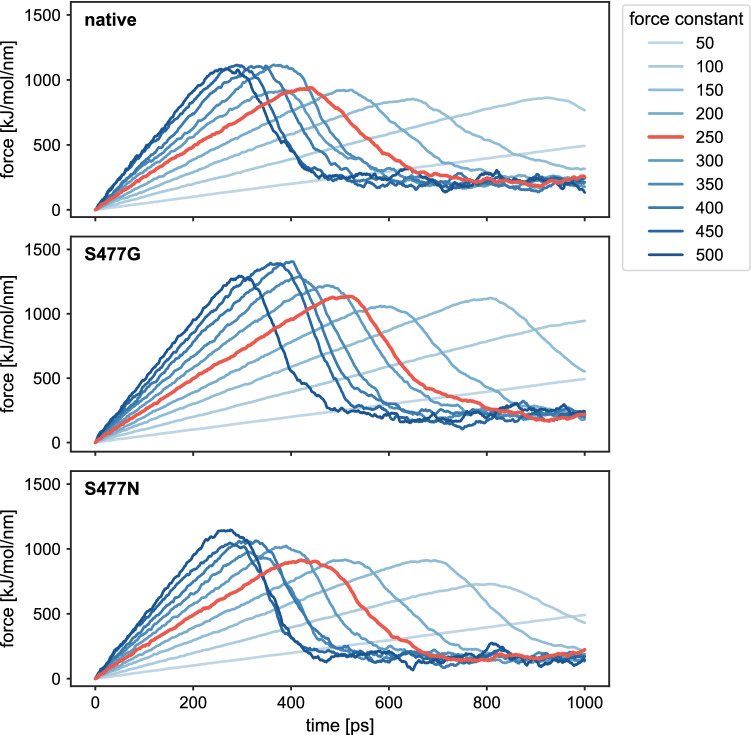
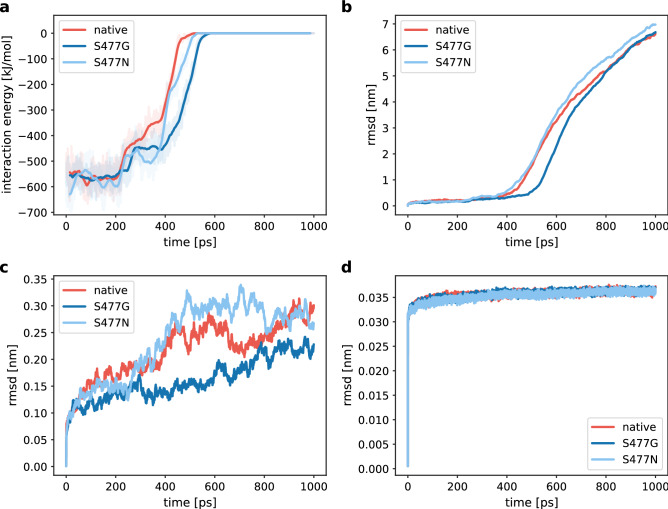
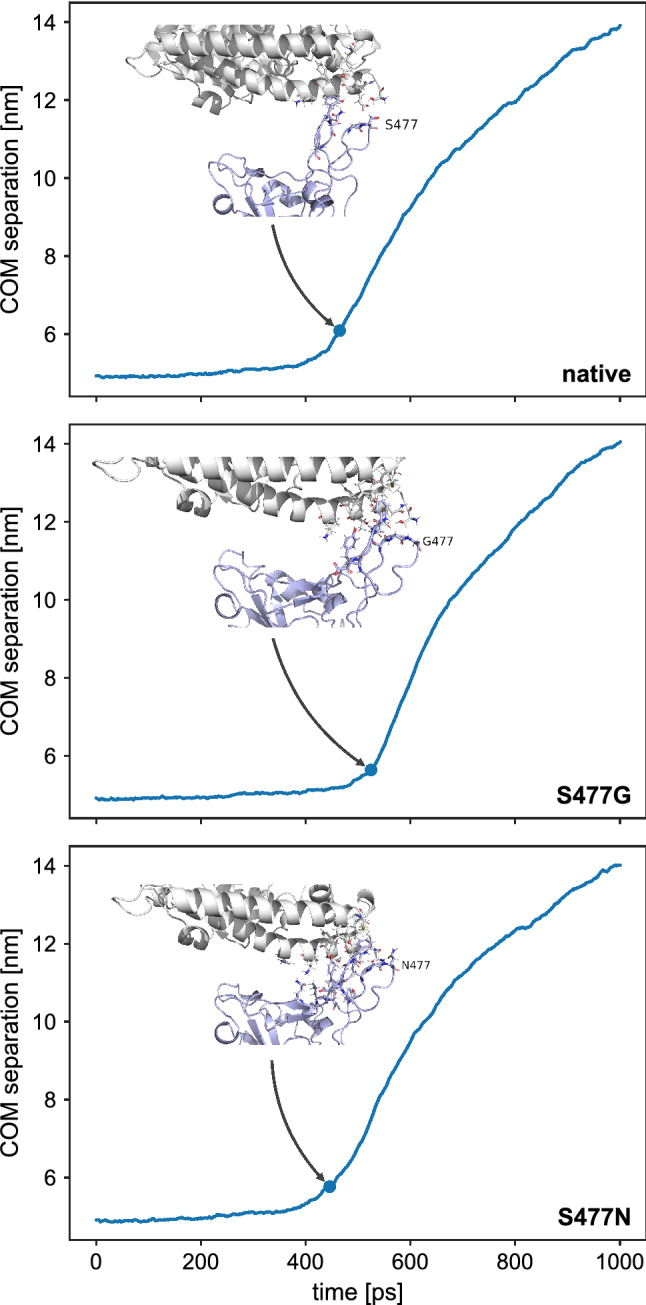
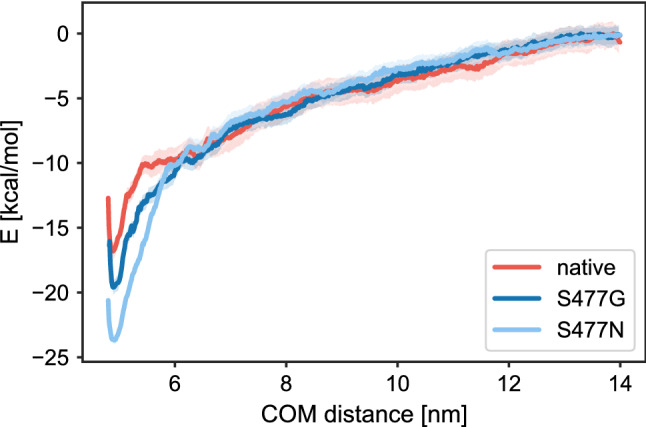
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
