

This is a preprint.
Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19
- PMID: 33619488
- PMCID: PMC7899452
- DOI: 10.1101/2021.02.20.431155
Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19
Update in
-
Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19.Cell. 2021 Sep 2;184(18):4713-4733.e22. doi: 10.1016/j.cell.2021.07.023. Epub 2021 Jul 23. Cell. 2021. PMID: 34352228 Free PMC article.
Abstract
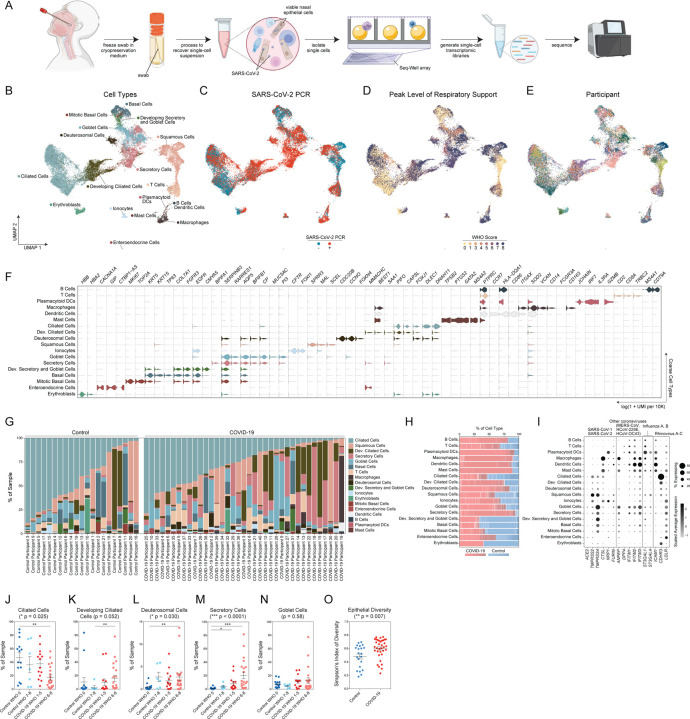
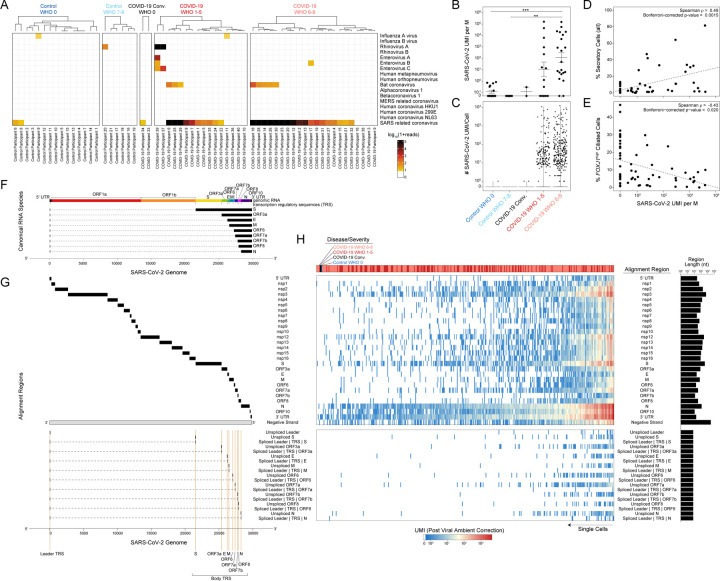
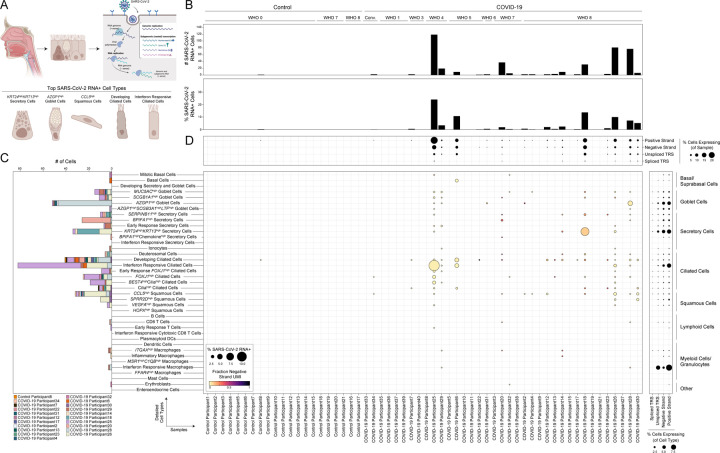
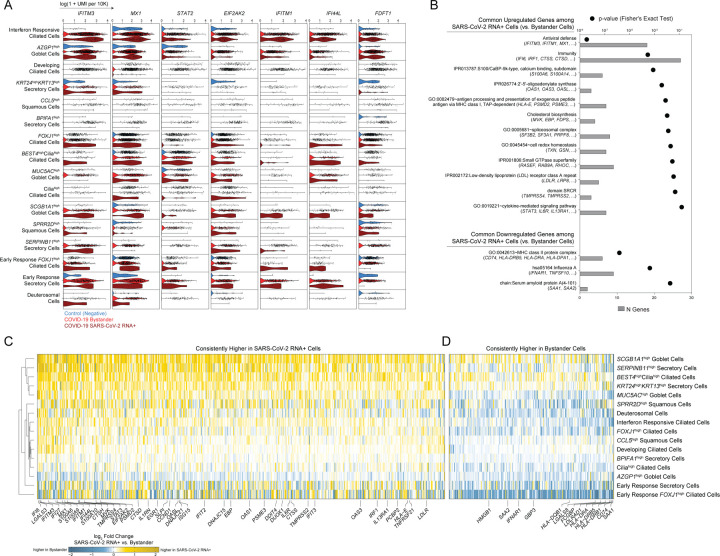
Infection with SARS-CoV-2, the virus that causes COVID-19, can lead to severe lower respiratory illness including pneumonia and acute respiratory distress syndrome, which can result in profound morbidity and mortality. However, many infected individuals are either asymptomatic or have isolated upper respiratory symptoms, which suggests that the upper airways represent the initial site of viral infection, and that some individuals are able to largely constrain viral pathology to the nasal and oropharyngeal tissues. Which cell types in the human nasopharynx are the primary targets of SARS-CoV-2 infection, and how infection influences the cellular organization of the respiratory epithelium remains incompletely understood. Here, we present nasopharyngeal samples from a cohort of 35 individuals with COVID-19, representing a wide spectrum of disease states from ambulatory to critically ill, as well as 23 healthy and intubated patients without COVID-19. Using standard nasopharyngeal swabs, we collected viable cells and performed single-cell RNA-sequencing (scRNA-seq), simultaneously profiling both host and viral RNA. We find that following infection with SARS-CoV-2, the upper respiratory epithelium undergoes massive reorganization: secretory cells diversify and expand, and mature epithelial cells are preferentially lost. Further, we observe evidence for deuterosomal cell and immature ciliated cell expansion, potentially representing active repopulation of lost ciliated cells through coupled secretory cell differentiation. Epithelial cells from participants with mild/moderate COVID-19 show extensive induction of genes associated with anti-viral and type I interferon responses. In contrast, cells from participants with severe lower respiratory symptoms appear globally muted in their anti-viral capacity, despite substantially higher local inflammatory myeloid populations and equivalent nasal viral loads. This suggests an essential role for intrinsic, local epithelial immunity in curbing and constraining viral-induced pathology. Using a custom computational pipeline, we characterized cell-associated SARS-CoV-2 RNA and identified rare cells with RNA intermediates strongly suggestive of active replication. Both within and across individuals, we find remarkable diversity and heterogeneity among SARS-CoV-2 RNA+ host cells, including developing/immature and interferon-responsive ciliated cells, KRT13+ "hillock"-like cells, and unique subsets of secretory, goblet, and squamous cells. Finally, SARS-CoV-2 RNA+ cells, as compared to uninfected bystanders, are enriched for genes involved in susceptibility (e.g., CTSL, TMPRSS2) or response (e.g., MX1, IFITM3, EIF2AK2) to infection. Together, this work defines both protective and detrimental host responses to SARS-CoV-2, determines the direct viral targets of infection, and suggests that failed anti-viral epithelial immunity in the nasal mucosa may underlie the progression to severe COVID-19.
Conflict of interest statement
DECLARATION OF INTERESTS A.K.S. reports compensation for consulting and/or SAB membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Hovione, Ochre Bio, and Dahlia Biosciences. J.O.M. reports compensation for consulting services with Cellarity and Hovione.
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous