

Virome composition in marine fish revealed by meta-transcriptomics
- PMID: 33623709
- PMCID: PMC7887440
- DOI: 10.1093/ve/veab005
Virome composition in marine fish revealed by meta-transcriptomics
Erratum in
-
Erratum: Virome composition in marine fish revealed by meta-transcriptomics.Virus Evol. 2021 Jun 17;7(1):veab035. doi: 10.1093/ve/veab035. eCollection 2021 Jan. Virus Evol. 2021. PMID: 34158971 Free PMC article.
Abstract
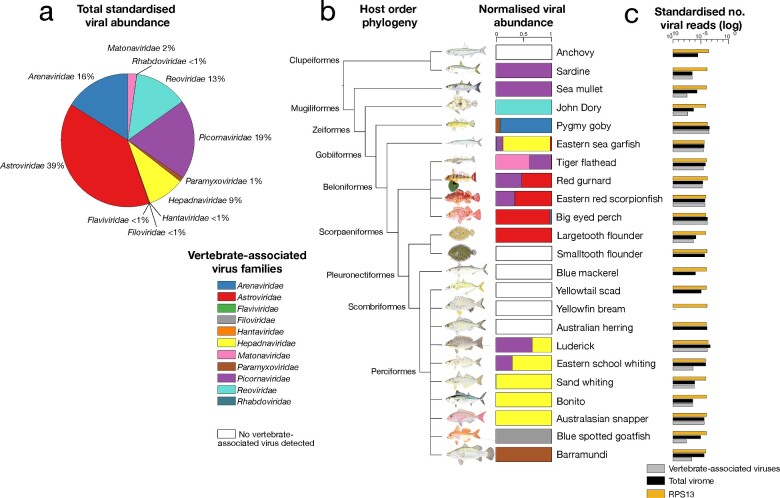
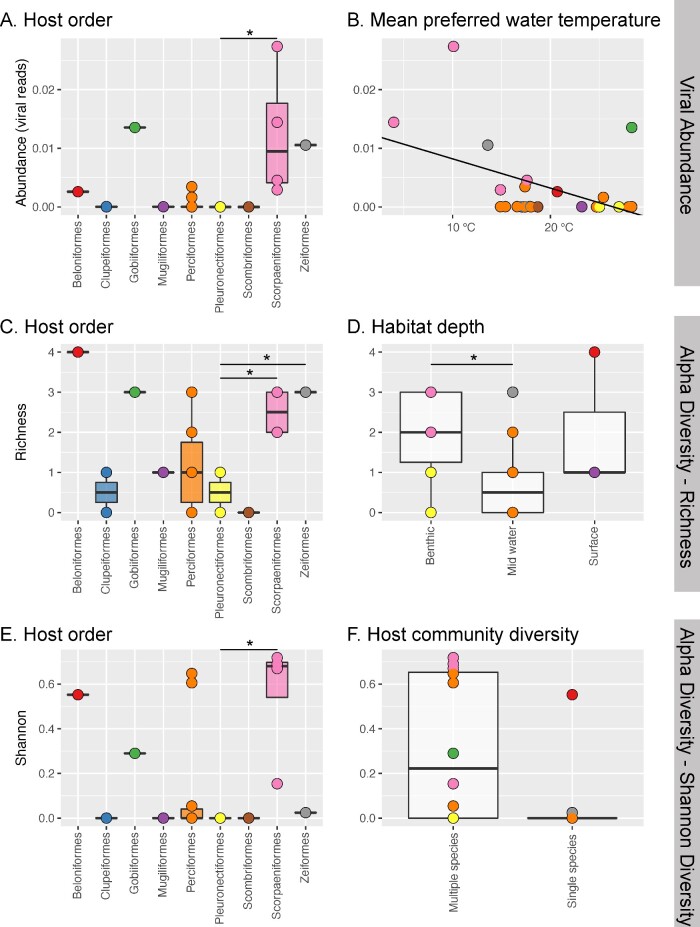
Revealing the determinants of virome composition is central to placing disease emergence in a broader evolutionary context. Fish are the most species-rich group of vertebrates and so provide an ideal model system to study the factors that shape virome compositions and their evolution. We characterized the viromes of nineteen wild-caught species of marine fish using total RNA sequencing (meta-transcriptomics) combined with analyses of sequence and protein structural homology to identify divergent viruses that often evade characterization. From this, we identified twenty-five new vertebrate-associated viruses and a further twenty-two viruses likely associated with fish diet or their microbiomes. The vertebrate-associated viruses identified here included the first fish virus in the Matonaviridae (single-strand, negative-sense RNA virus). Other viruses fell within the Astroviridae, Picornaviridae, Arenaviridae, Reoviridae, Hepadnaviridae, Paramyxoviridae, Rhabdoviridae, Hantaviridae, Filoviridae, and Flaviviridae, and were sometimes phylogenetically distinct from known fish viruses. We also show how key metrics of virome composition-viral richness, abundance, and diversity-can be analysed along with host ecological and biological factors as a means to understand virus ecology. Accordingly, these data suggest that that the vertebrate-associated viromes of the fish sampled here are predominantly shaped by the phylogenetic history (i.e. taxonomic order) of their hosts, along with several biological factors including water temperature, habitat depth, community diversity and swimming behaviour. No such correlations were found for viruses associated with porifera, molluscs, arthropods, fungi, and algae, that are unlikely to replicate in fish hosts. Overall, these data indicate that fish harbour particularly large and complex viromes and the vast majority of fish viromes are undescribed.
Keywords: fish; host-jumping; metagenomics; virome; virus evolution.
© The Author(s) 2021. Published by Oxford University Press.
Figures

Similar articles
-
Metagenomic sequencing reveals a lack of virus exchange between native and invasive freshwater fish across the Murray-Darling Basin, Australia.Virus Evol. 2021 Apr 13;7(1):veab034. doi: 10.1093/ve/veab034. eCollection 2021 Jan. Virus Evol. 2021. PMID: 34017611 Free PMC article.
-
The diverse liver viromes of Australian geckos and skinks are dominated by hepaciviruses and picornaviruses and reflect host taxonomy and habitat.Virus Evol. 2024 May 28;10(1):veae044. doi: 10.1093/ve/veae044. eCollection 2024. Virus Evol. 2024. PMID: 38854849 Free PMC article.
-
Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition.Viruses. 2022 Jan 27;14(2):257. doi: 10.3390/v14020257. Viruses. 2022. PMID: 35215850 Free PMC article.
-
Making sense of the virome in light of evolution and ecology.Proc Biol Sci. 2025 Apr;292(2044):20250389. doi: 10.1098/rspb.2025.0389. Epub 2025 Apr 2. Proc Biol Sci. 2025. PMID: 40169018 Free PMC article. Review.
-
Diversity, evolution, and emergence of fish viruses.J Virol. 2024 Jun 13;98(6):e0011824. doi: 10.1128/jvi.00118-24. Epub 2024 May 24. J Virol. 2024. PMID: 38785422 Free PMC article. Review.
Cited by
-
Virome Analysis of Normal and Growth Retardation Disease-Affected Macrobrachium rosenbergii.Microbiol Spectr. 2022 Dec 21;10(6):e0146222. doi: 10.1128/spectrum.01462-22. Epub 2022 Nov 29. Microbiol Spectr. 2022. PMID: 36445118 Free PMC article.
-
Identification of a novel filovirus in a common lancehead (Bothrops atrox (Linnaeus, 1758)).J Vet Med Sci. 2021 Sep 27;83(9):1485-1488. doi: 10.1292/jvms.21-0285. Epub 2021 Jul 19. J Vet Med Sci. 2021. PMID: 34275961 Free PMC article.
-
Exploring the Astrovirome of Shellfish Matrices Using Nanopore Sequencing.Vet Sci. 2023 Feb 21;10(3):175. doi: 10.3390/vetsci10030175. Vet Sci. 2023. PMID: 36977214 Free PMC article.
-
Novel filoviruses: indication of a global threat or cause to reassess our risk perception?Npj Viruses. 2024 Aug 21;2(1):38. doi: 10.1038/s44298-024-00050-4. Npj Viruses. 2024. PMID: 40295872 Free PMC article. Review.
-
Altered vaginal eukaryotic virome is associated with different cervical disease status.Virol Sin. 2023 Apr;38(2):184-197. doi: 10.1016/j.virs.2022.12.004. Epub 2022 Dec 21. Virol Sin. 2023. PMID: 36565811 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
