

Enzyme Replacement Therapy for Succinic Semialdehyde Dehydrogenase Deficiency: Relevance in γ-Aminobutyric Acid Plasticity
- PMID: 33624531
- PMCID: PMC8382780
- DOI: 10.1177/0883073821993000
Enzyme Replacement Therapy for Succinic Semialdehyde Dehydrogenase Deficiency: Relevance in γ-Aminobutyric Acid Plasticity
Abstract
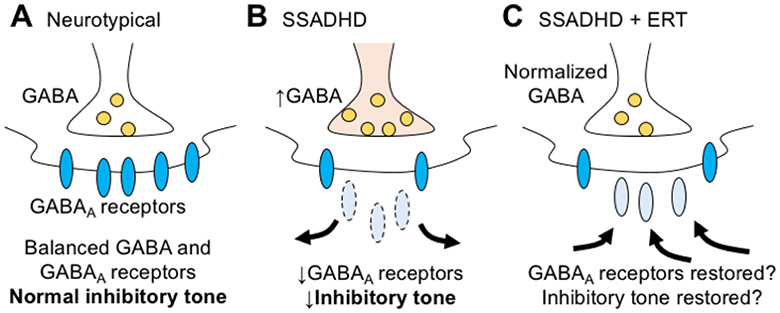
Succinic semialdehyde dehydrogenase deficiency (SSADHD) is a rare inborn metabolic disorder caused by the functional impairment of SSADH (encoded by the ALDH5A1 gene), an enzyme essential for metabolism of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). In SSADHD, pathologic accumulation of GABA and its metabolite γ-hydroxybutyrate (GHB) results in broad spectrum encephalopathy including developmental delay, ataxia, seizures, and a heightened risk of sudden unexpected death in epilepsy (SUDEP). Proof-of-concept systemic SSADH restoration via enzyme replacement therapy increased survival of SSADH knockout mice, suggesting that SSADH restoration might be a viable intervention for SSADHD. However, before testing enzyme replacement therapy or gene therapy in patients, we must consider its safety and feasibility in the context of early brain development and unique SSADHD pathophysiology. Specifically, a profound use-dependent downregulation of GABAA receptors in SSADHD indicates a risk that any sudden SSADH restoration might diminish GABAergic tone and provoke seizures. In addition, the tight developmental regulation of GABA circuit plasticity might limit the age window when SSADH restoration is accomplished safely. Moreover, given SSADH expressions are cell type-specific, targeted instead of global restoration might be necessary. We therefore describe 3 key parameters for the clinical readiness of SSADH restoration: (1) rate, (2) timing, and (3) cell type specificity. Our work focuses on the construction of a novel SSADHD mouse model that allows "on-demand" SSADH restoration for the systematic investigation of these key parameters. We aim to understand the impacts of specific SSADH restoration protocols on brain physiology, accelerating bench-to-bedside development of enzyme replacement therapy or gene therapy for SSADHD patients.
Keywords: epilepsy; genetics; inborn errors of metabolism; metabolism; mitochondrial disorder; neurodevelopment; seizures; status epilepticus; treatment.
Figures








References
-
- Pearl PL, Wiwattanadittakul N, Roullet JB & Gibson KM in GeneReviews((R)) (eds Adam MP et al.) (1993).
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
