

Multi-kingdom ecological drivers of microbiota assembly in preterm infants
- PMID: 33627867
- PMCID: PMC7990694
- DOI: 10.1038/s41586-021-03241-8
Multi-kingdom ecological drivers of microbiota assembly in preterm infants
Abstract
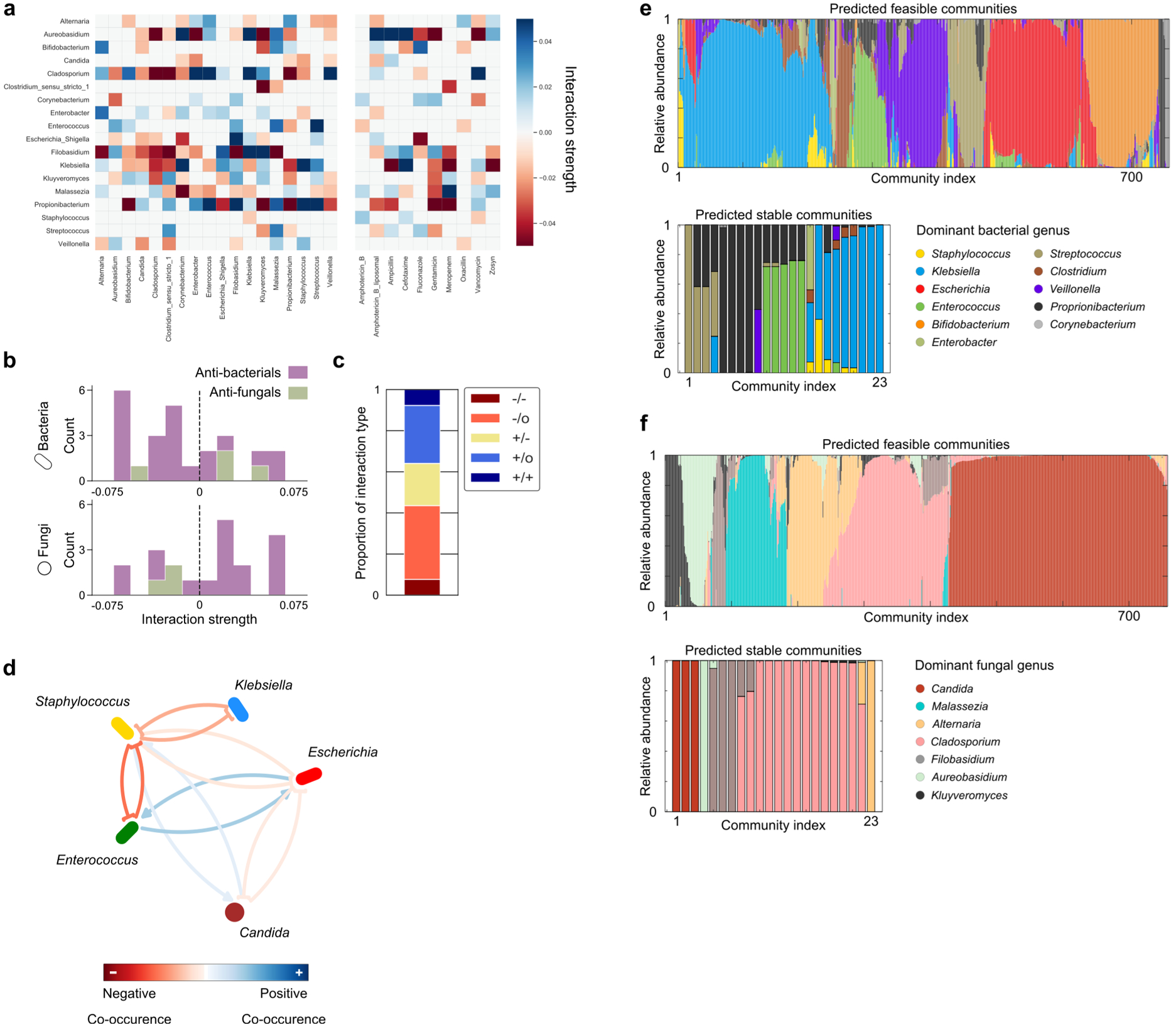
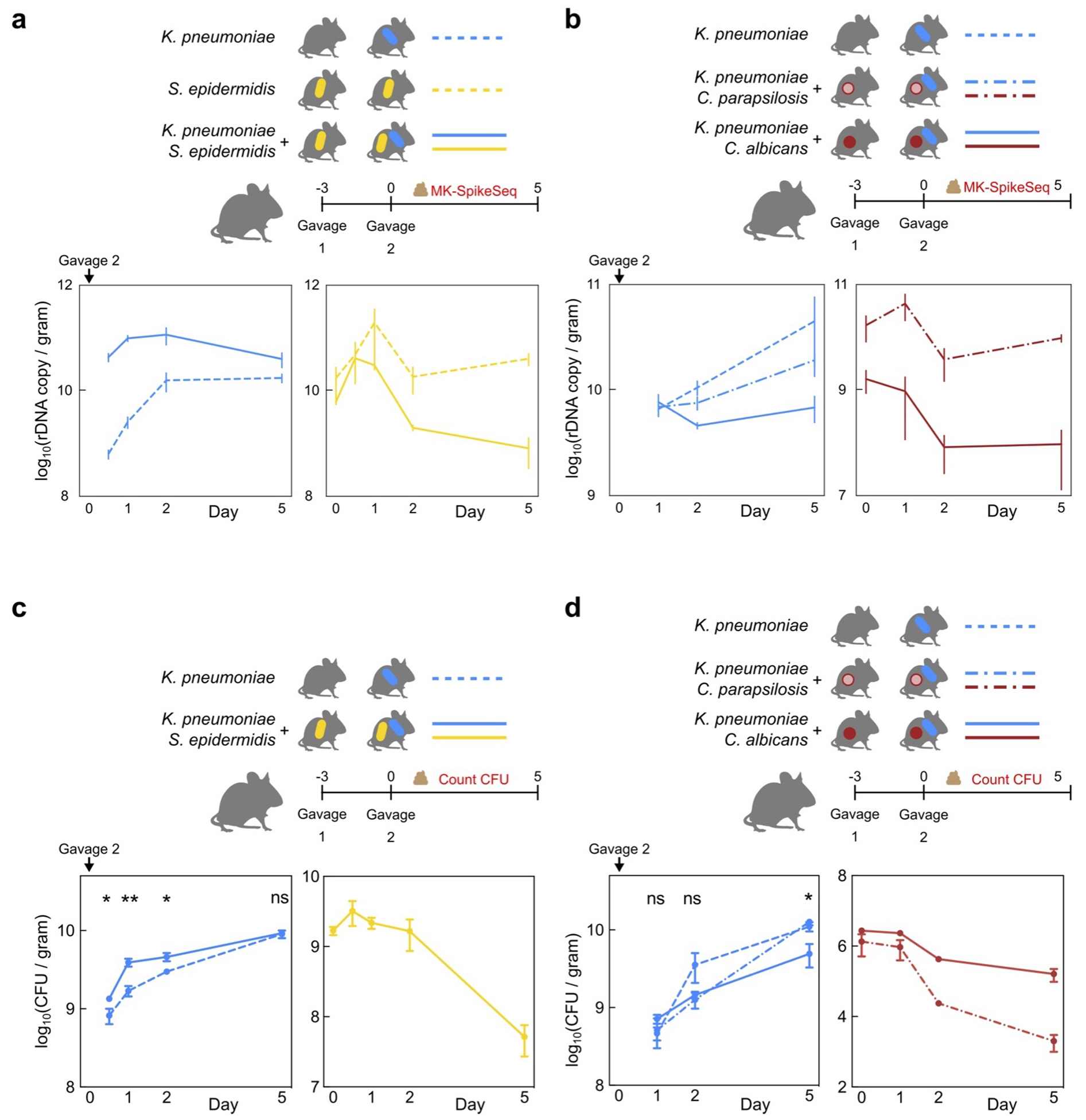
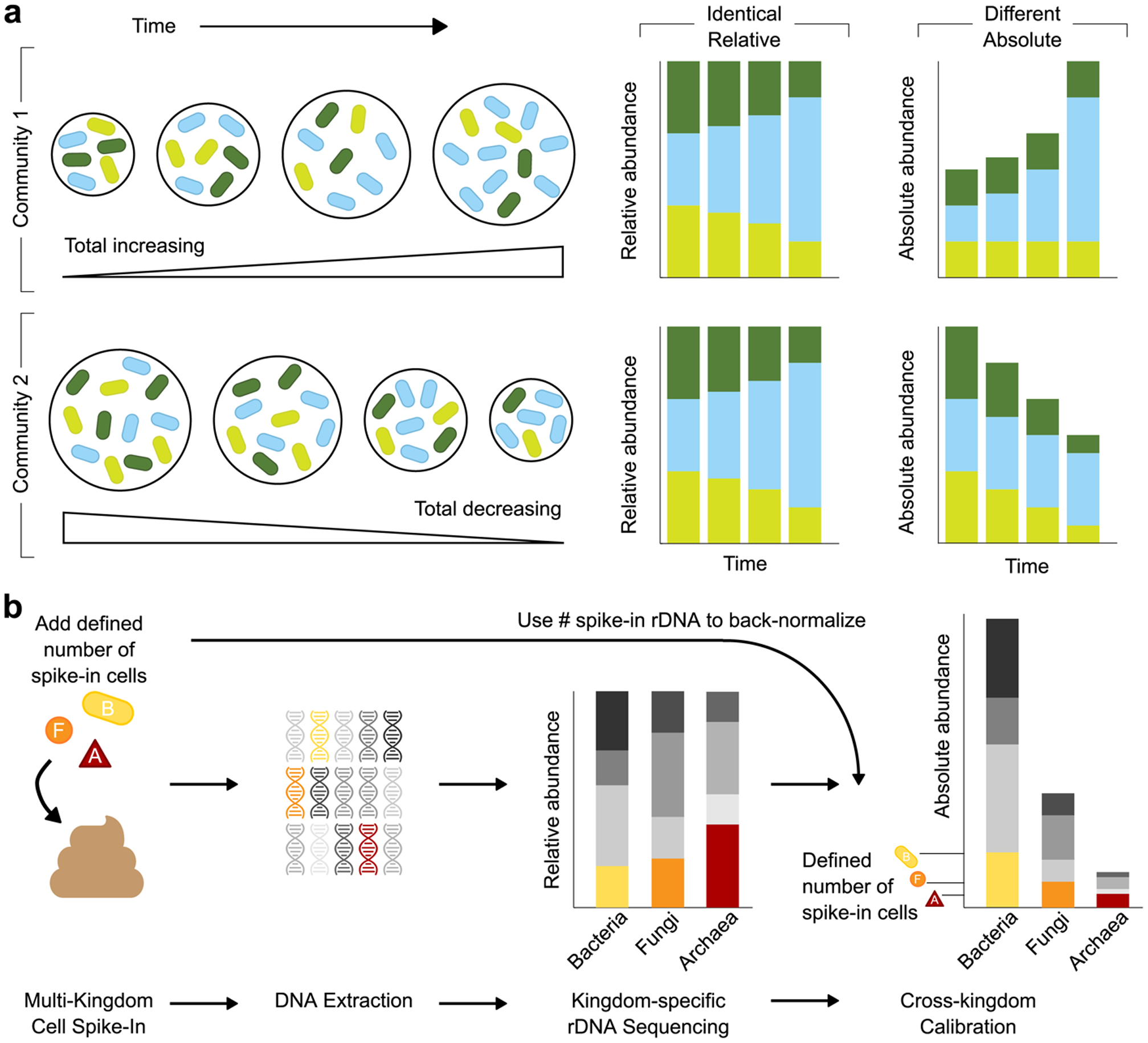
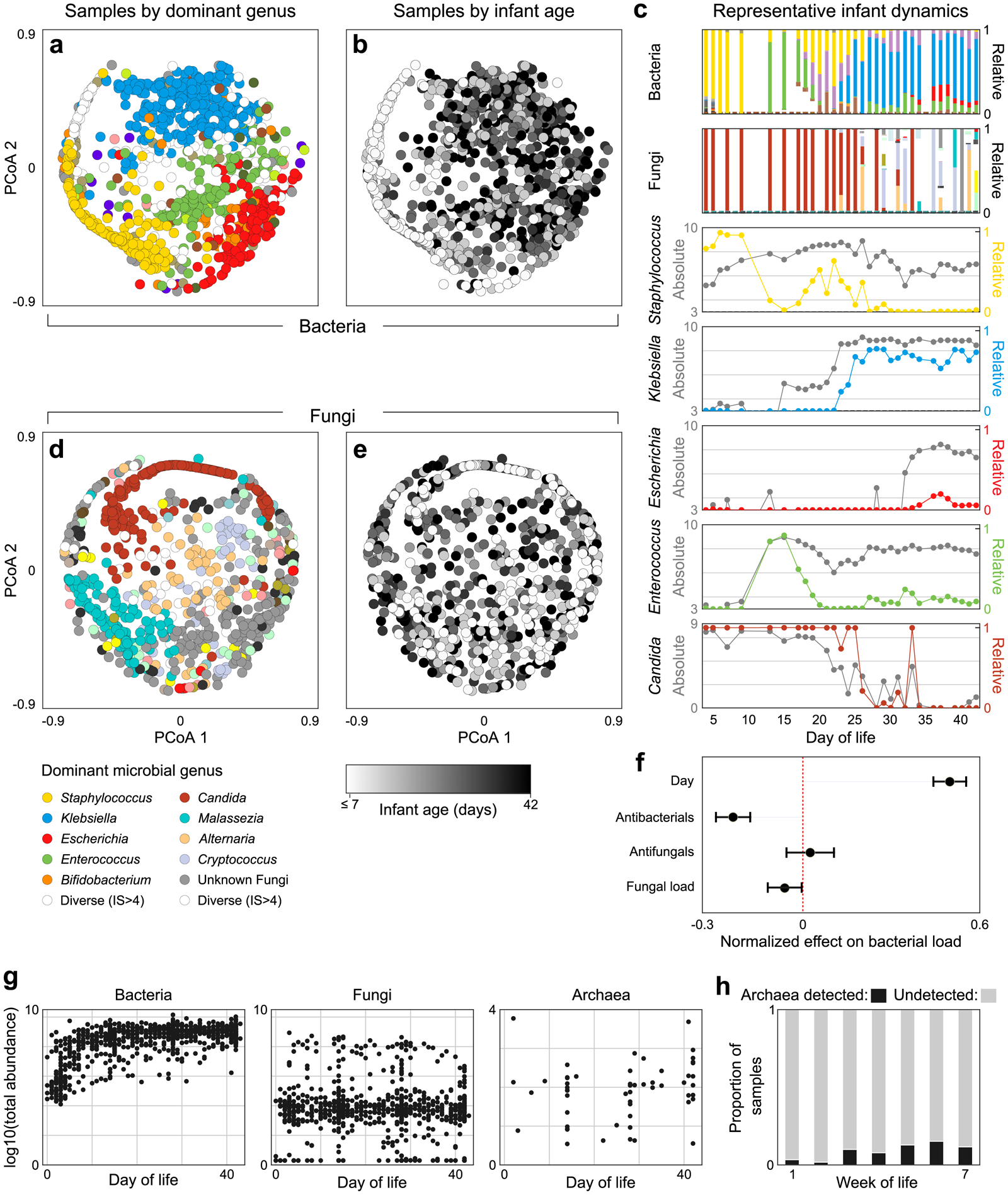
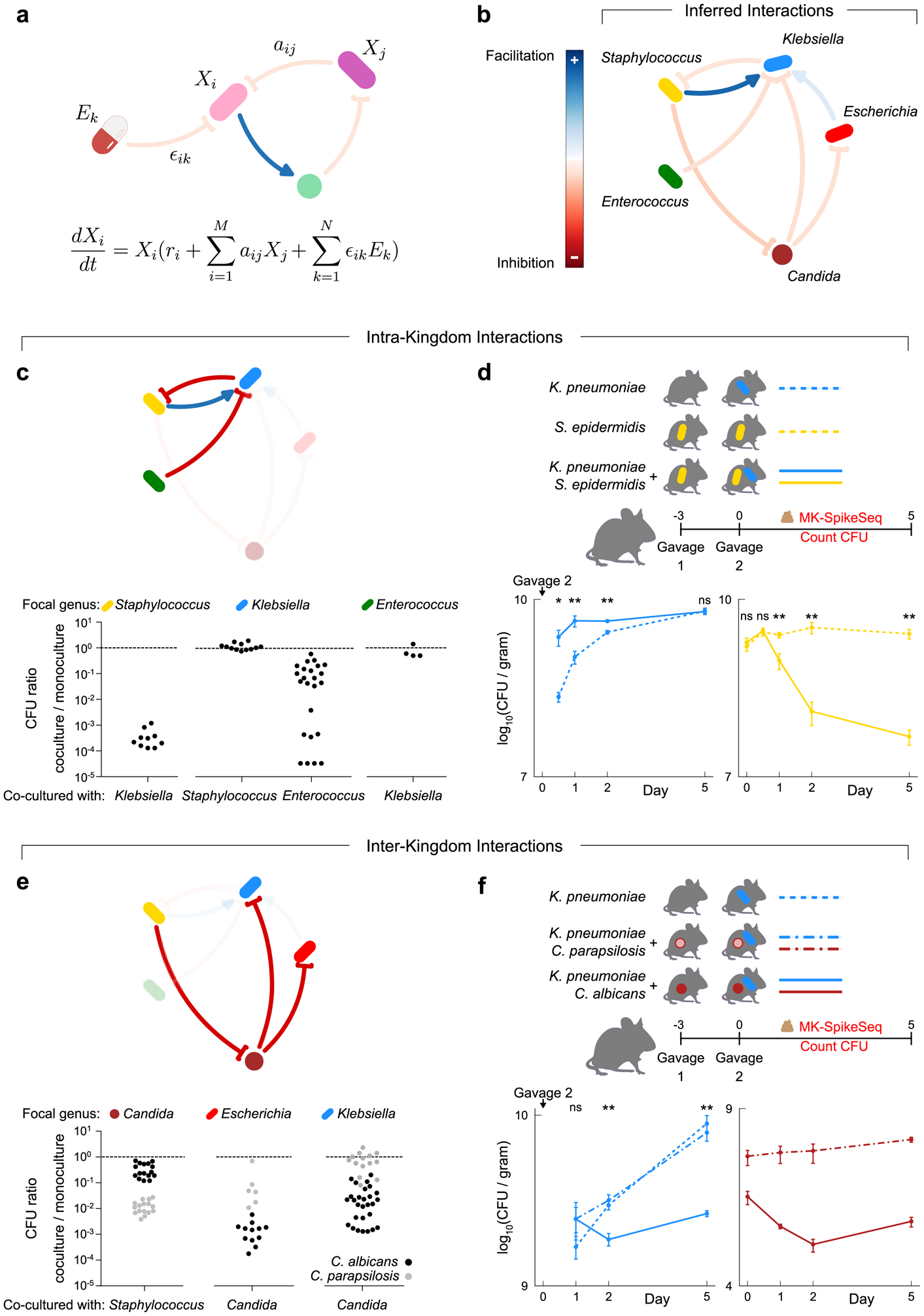
The gut microbiota of preterm infants develops predictably1-7, with pioneer species colonizing the gut after birth, followed by an ordered succession of microorganisms. The gut microbiota is vital to the health of preterm infants8,9, but the forces that shape these predictable dynamics of microbiome assembly are unknown. The environment, the host and interactions between microorganisms all potentially shape the dynamics of the microbiota, but in such a complex ecosystem, identifying the specific role of any individual factor is challenging10-14. Here we use multi-kingdom absolute abundance quantification, ecological modelling and experimental validation to address this challenge. We quantify the absolute dynamics of bacteria, fungi and archaea in a longitudinal cohort of 178 preterm infants. We uncover microbial blooms and extinctions, and show that there is an inverse correlation between bacterial and fungal loads in the infant gut. We infer computationally and demonstrate experimentally in vitro and in vivo that predictable assembly dynamics may be driven by directed, context-dependent interactions between specific microorganisms. Mirroring the dynamics of macroscopic ecosystems15-17, a late-arriving member of the microbiome, Klebsiella, exploits the pioneer microorganism, Staphylococcus, to gain a foothold within the gut. Notably, we find that interactions between different kingdoms can influence assembly, with a single fungal species-Candida albicans-inhibiting multiple dominant genera of gut bacteria. Our work reveals the centrality of simple microbe-microbe interactions in shaping host-associated microbiota, which is critical both for our understanding of microbiota ecology and for targeted microbiota interventions.
Conflict of interest statement
Competing interests
CRM receives grant funding from Mead Johnson Nutrition. CRM also provides consulting services for Mead Johnson Nutrition, Alcresta and Fresenius Kabi, and sits on the Scientific Advisory Boards of Plakous Therapeutics and LUCA Biologics. All other authors declare no competing interests.
Figures
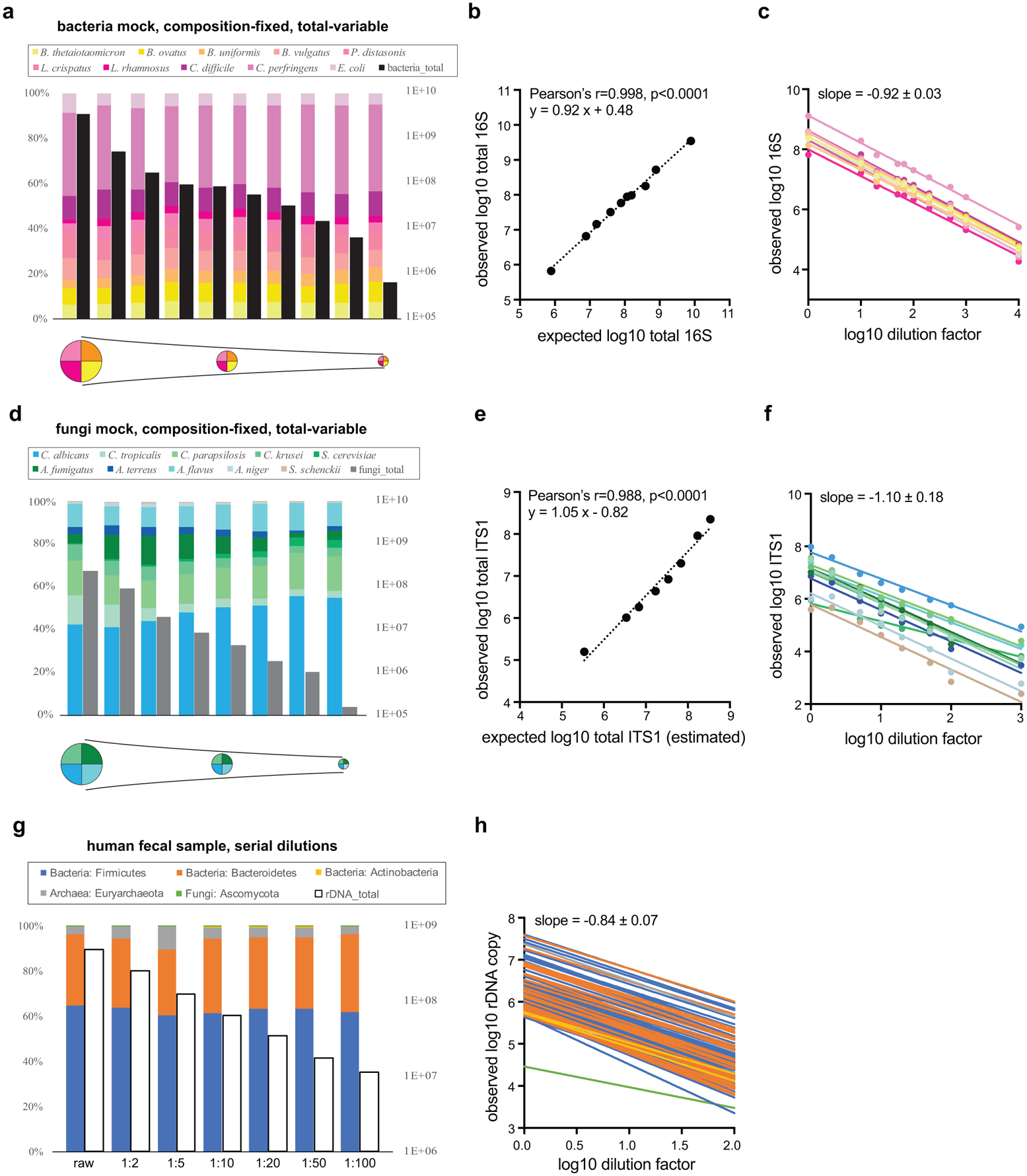
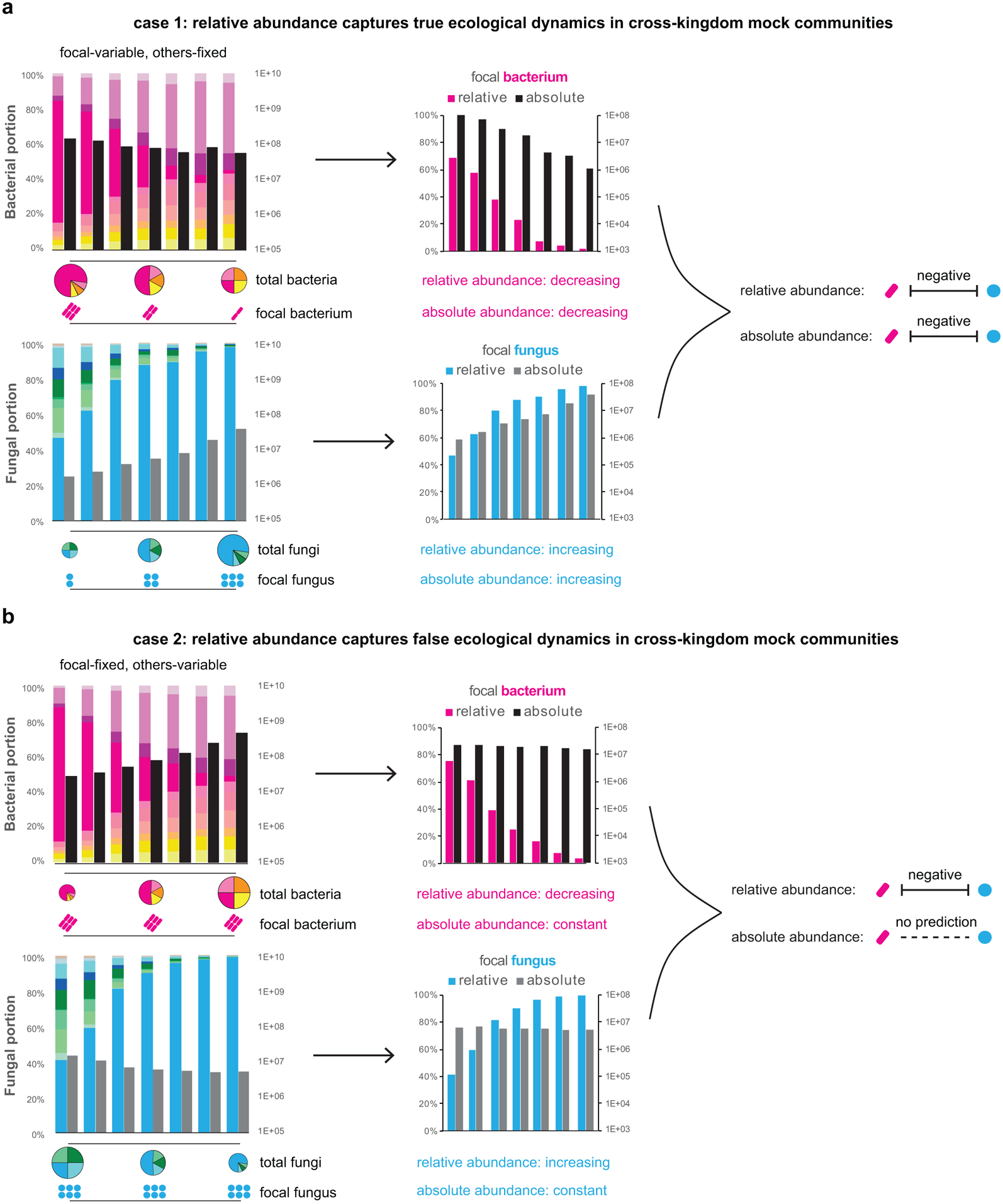
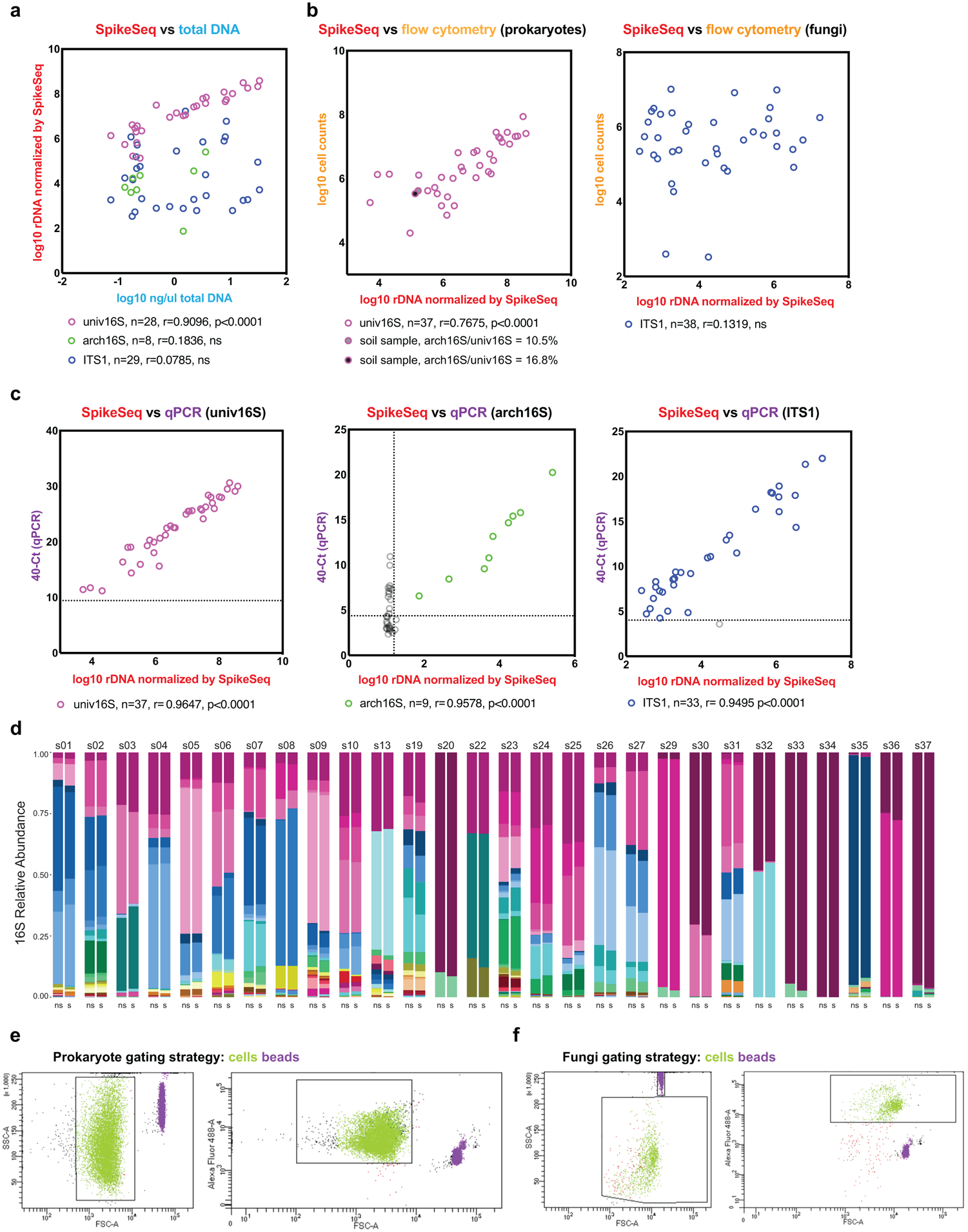
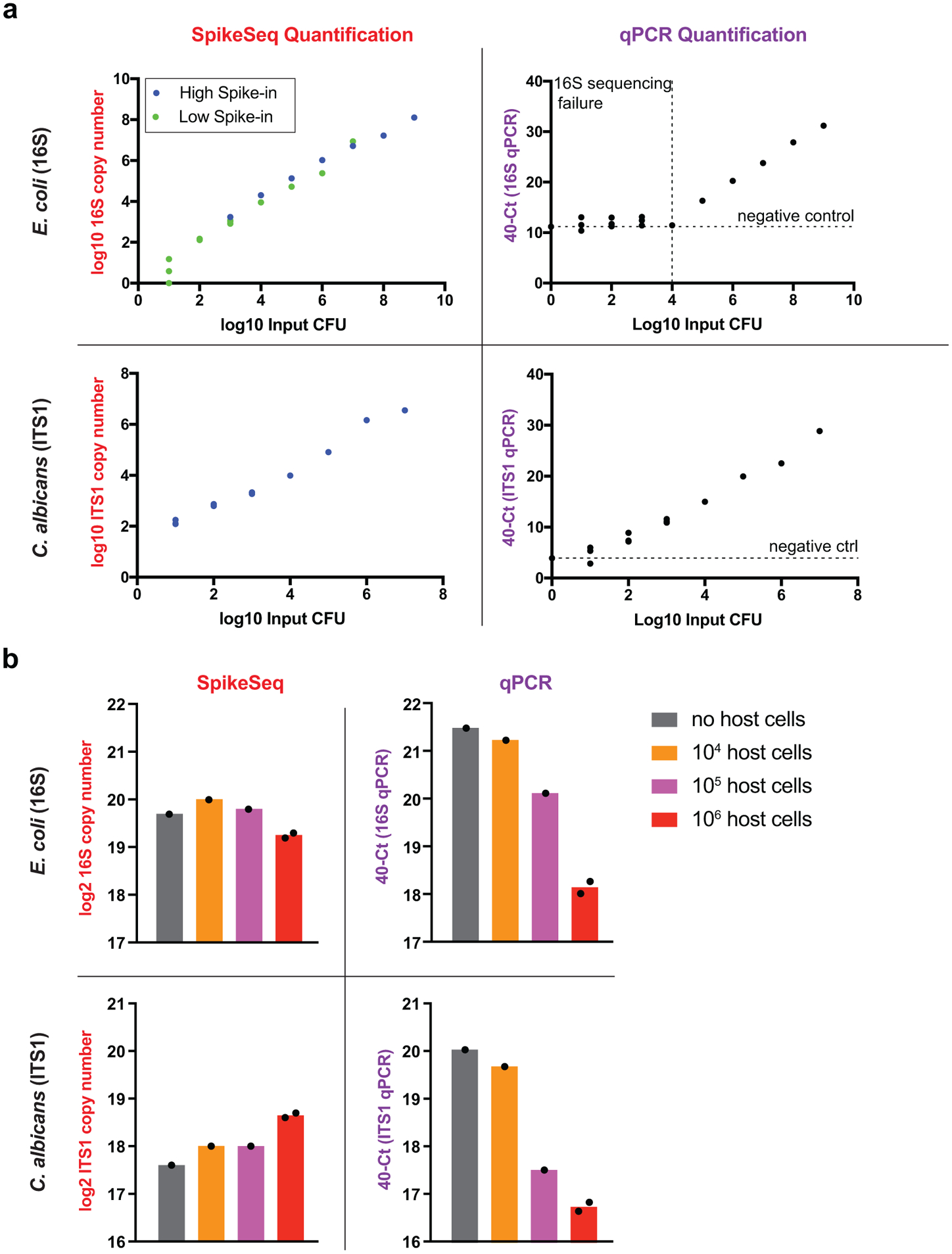
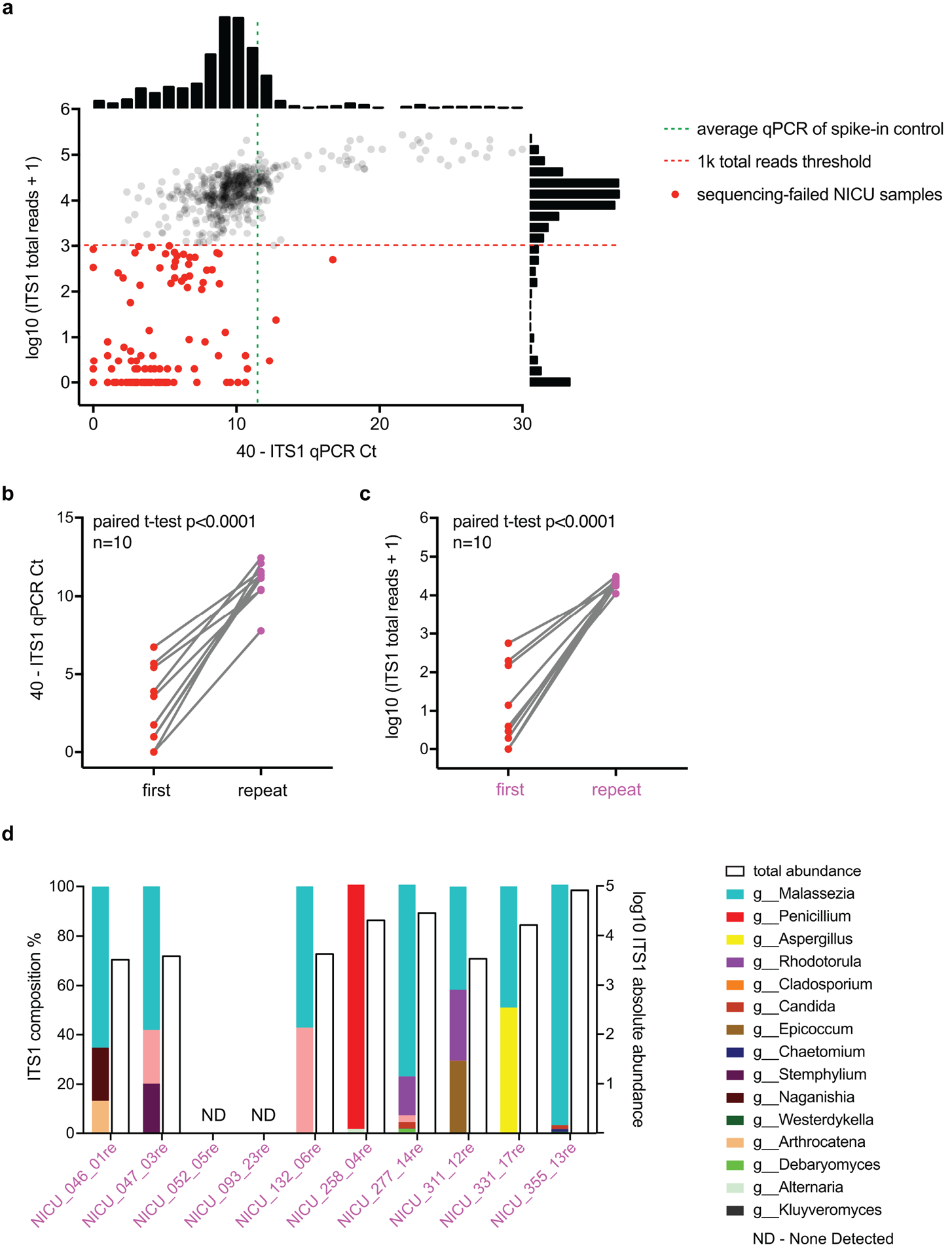
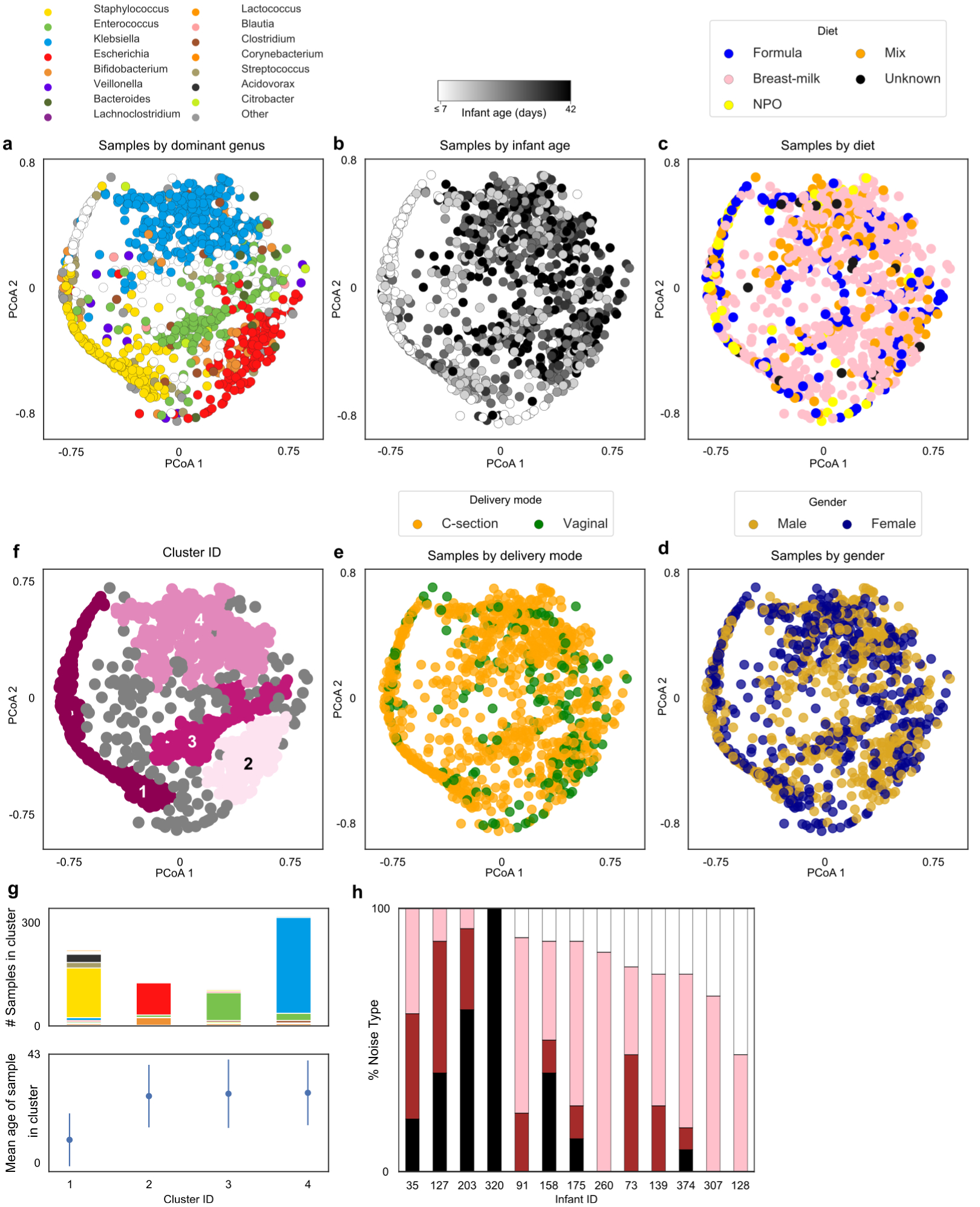
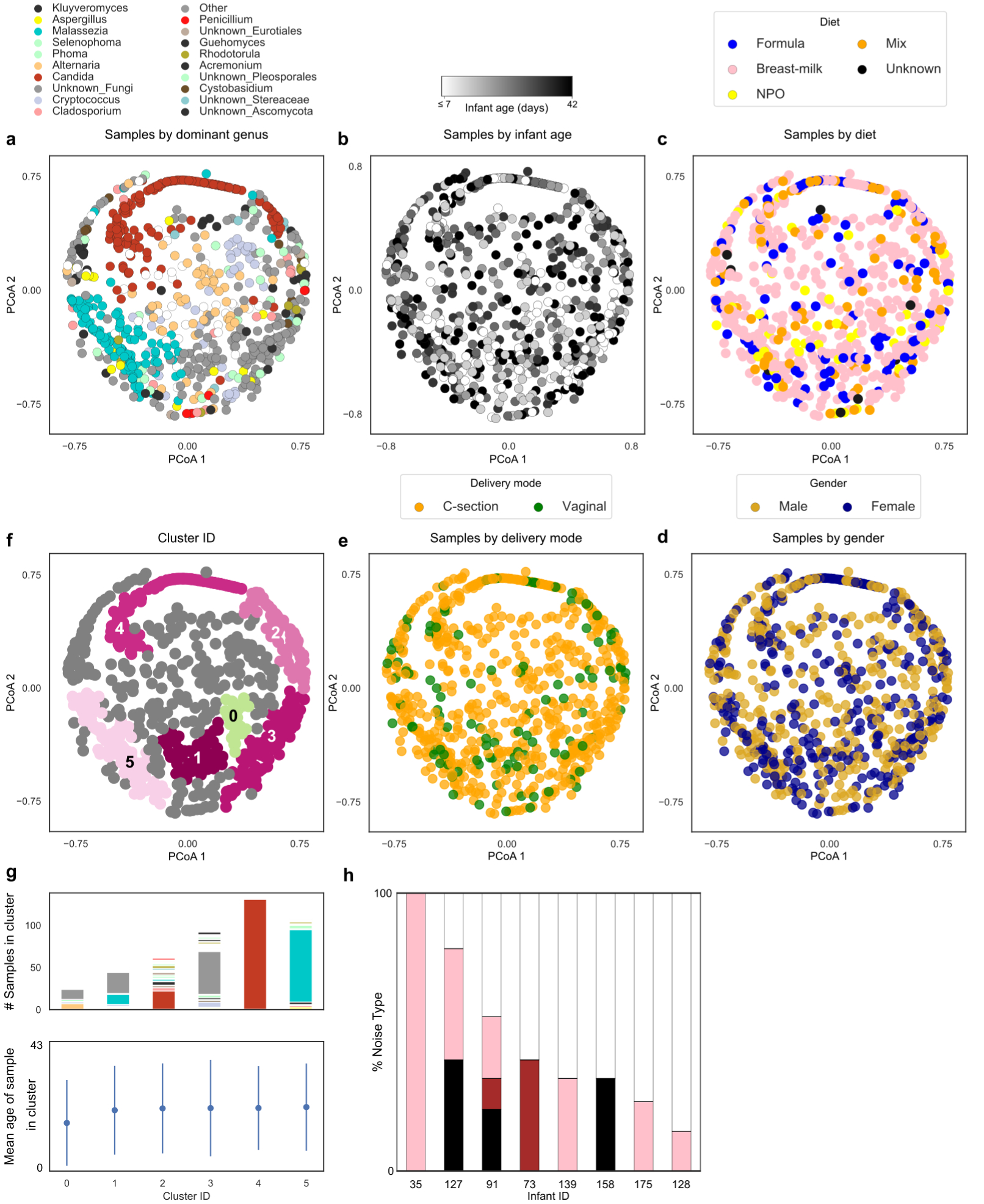
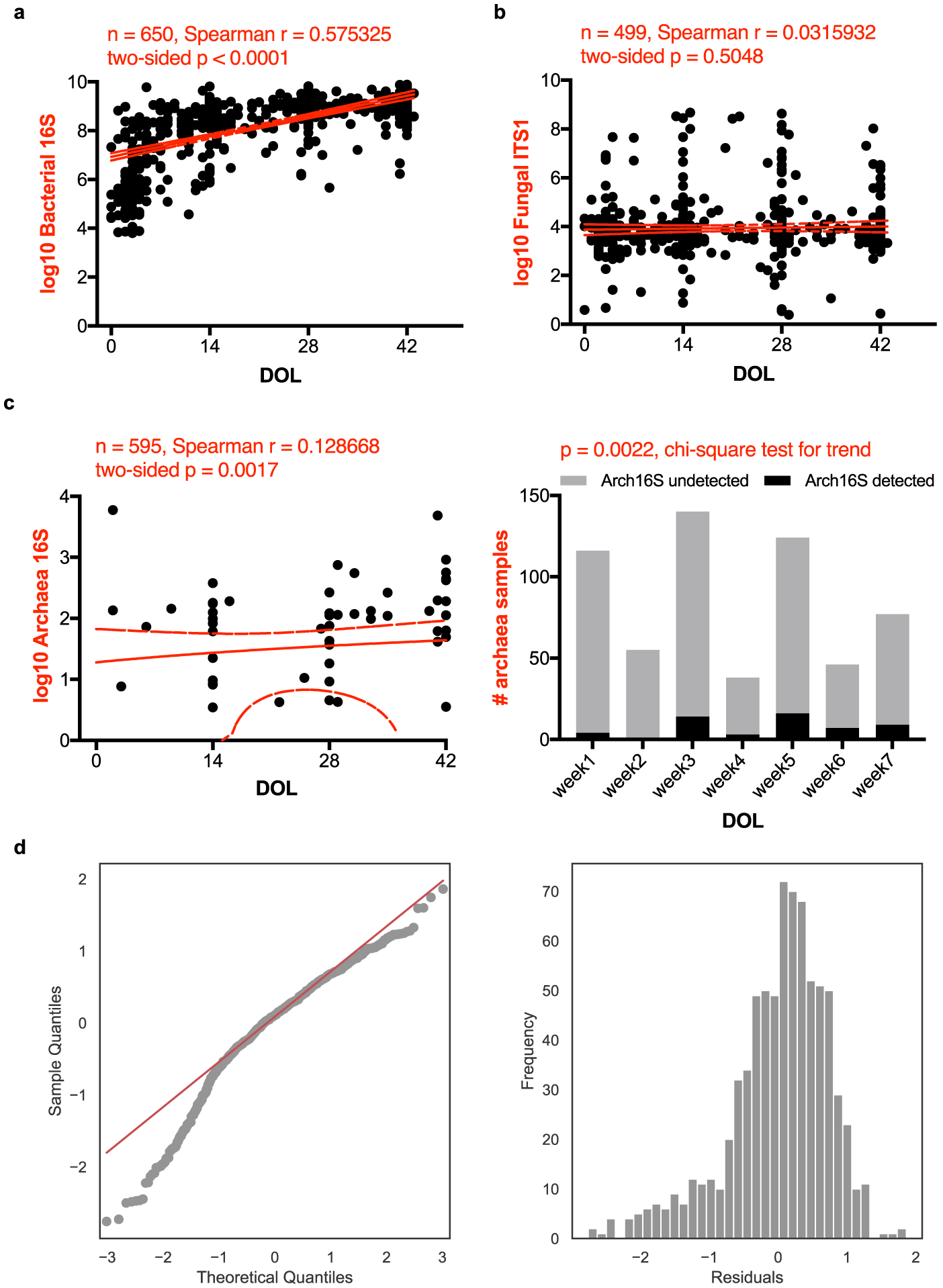
Comment in
-
Inter-microbial relations shape the preterm gut.Nat Microbiol. 2021 Apr;6(4):423-424. doi: 10.1038/s41564-021-00883-2. Nat Microbiol. 2021. PMID: 33753920 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
