

Bardet-Biedl syndrome proteins regulate intracellular signaling and neuronal function in patient-specific iPSC-derived neurons
- PMID: 33630762
- PMCID: PMC8262481
- DOI: 10.1172/JCI146287
Bardet-Biedl syndrome proteins regulate intracellular signaling and neuronal function in patient-specific iPSC-derived neurons
Abstract
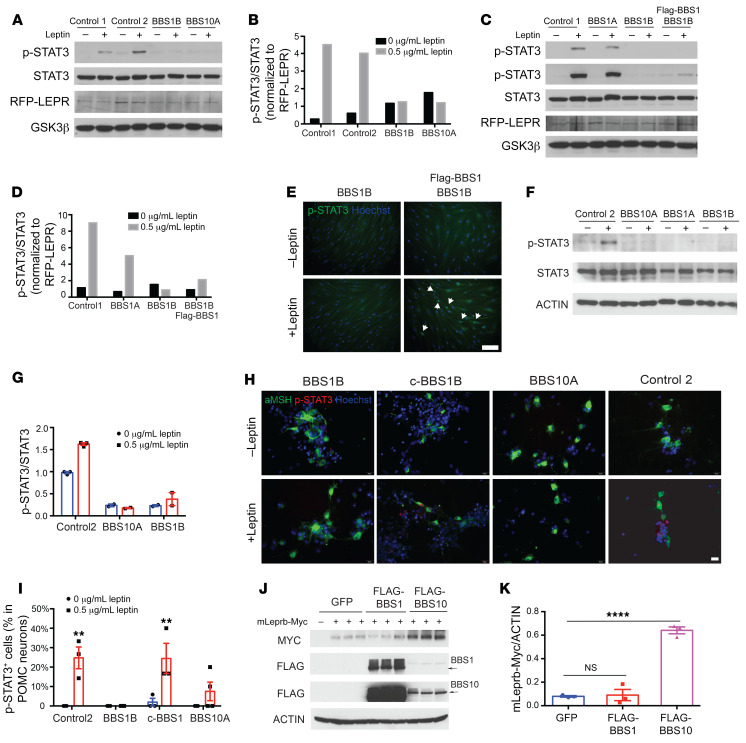
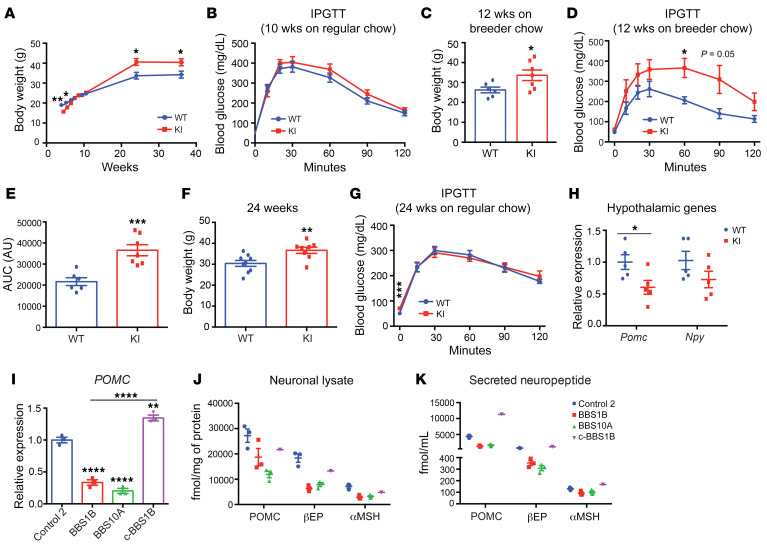
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disorder caused by mutations in genes encoding components of the primary cilium and is characterized by hyperphagic obesity. To investigate the molecular basis of obesity in human BBS, we developed a cellular model of BBS using induced pluripotent stem cell-derived (iPSC-derived) hypothalamic arcuate-like neurons. BBS mutations BBS1M390R and BBS10C91fsX95 did not affect neuronal differentiation efficiency but caused morphological defects, including impaired neurite outgrowth and longer primary cilia. Single-cell RNA sequencing of BBS1M390R hypothalamic neurons identified several downregulated pathways, including insulin and cAMP signaling and axon guidance. Additional studies demonstrated that BBS1M390R and BBS10C91fsX95 mutations impaired insulin signaling in both human fibroblasts and iPSC-derived neurons. Overexpression of intact BBS10 fully restored insulin signaling by restoring insulin receptor tyrosine phosphorylation in BBS10C91fsX95 neurons. Moreover, mutations in BBS1 and BBS10 impaired leptin-mediated p-STAT3 activation in iPSC-derived hypothalamic neurons. Correction of the BBS mutation by CRISPR rescued leptin signaling. POMC expression and neuropeptide production were decreased in BBS1M390R and BBS10C91fsX95 iPSC-derived hypothalamic neurons. In the aggregate, these data provide insights into the anatomic and functional mechanisms by which components of the BBSome in CNS primary cilia mediate effects on energy homeostasis.
Keywords: Obesity; Stem cells.
Conflict of interest statement
Figures
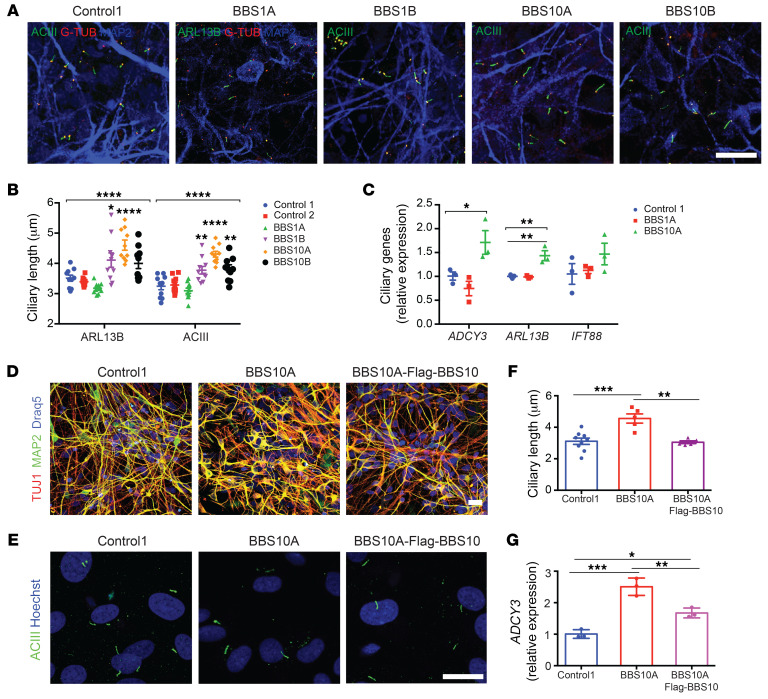
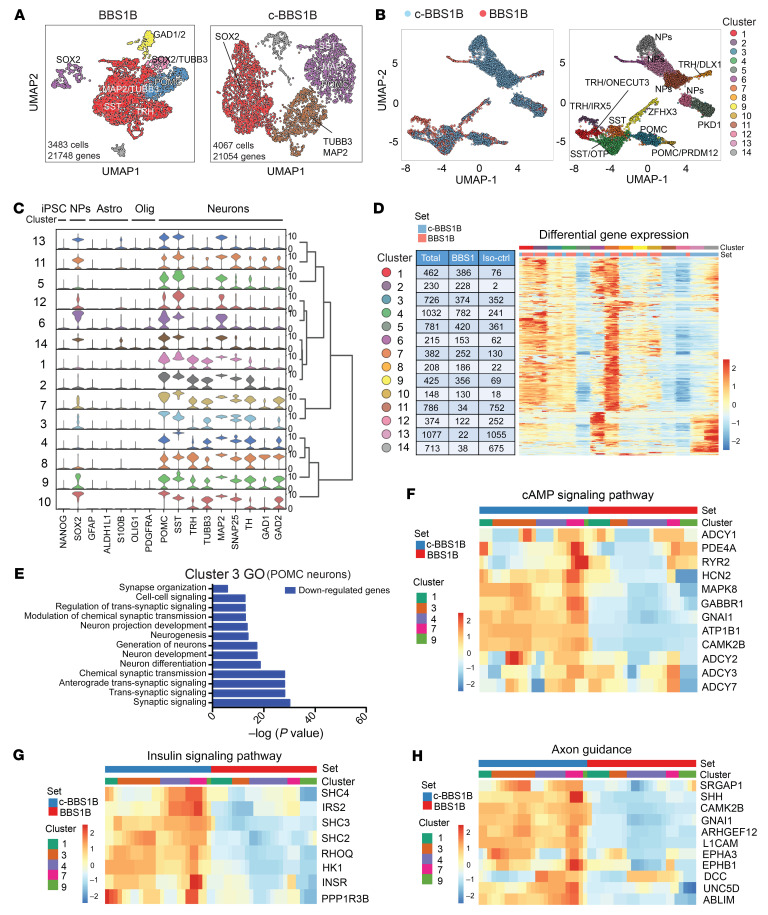
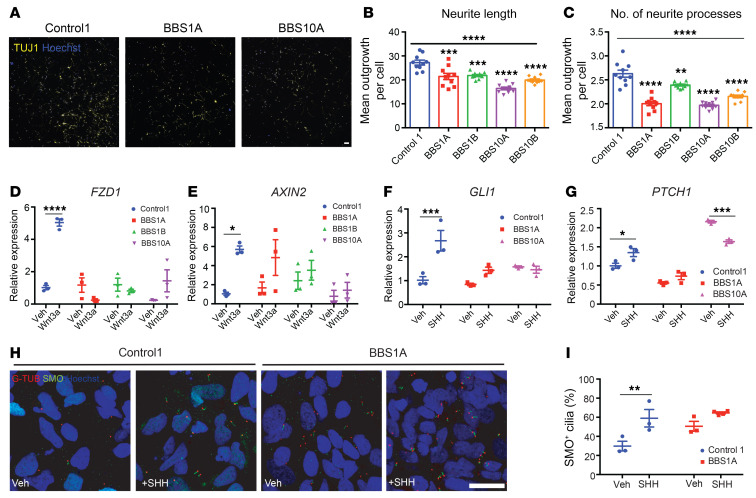
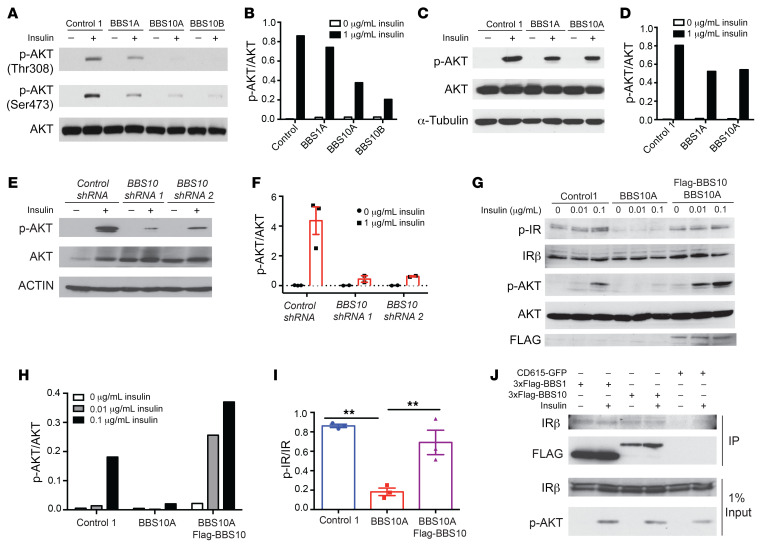
Comment in
-
The BBSome: a nexus controlling energy metabolism in the brain.J Clin Invest. 2021 Apr 15;131(8):e148903. doi: 10.1172/JCI148903. J Clin Invest. 2021. PMID: 33855975 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- R21 AG050437/AG/NIA NIH HHS/United States
- R01 EY018213/EY/NEI NIH HHS/United States
- R01 EY026682/EY/NEI NIH HHS/United States
- R24 EY027285/EY/NEI NIH HHS/United States
- R01 DK064819/DK/NIDDK NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- P30 DK026687/DK/NIDDK NIH HHS/United States
- P30 DK063608/DK/NIDDK NIH HHS/United States
- U01 EY030580/EY/NEI NIH HHS/United States
- K01 DK123199/DK/NIDDK NIH HHS/United States
- P50 HD105352/HD/NICHD NIH HHS/United States
- U54 OD020351/OD/NIH HHS/United States
- R01 EY024698/EY/NEI NIH HHS/United States
- P30 EY019007/EY/NEI NIH HHS/United States
- R01 DK052431/DK/NIDDK NIH HHS/United States
- R01 DK093920/DK/NIDDK NIH HHS/United States
- R24 EY028758/EY/NEI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
