

Impact of between-tissue differences on pan-cancer predictions of drug sensitivity
- PMID: 33630864
- PMCID: PMC7906305
- DOI: 10.1371/journal.pcbi.1008720
Impact of between-tissue differences on pan-cancer predictions of drug sensitivity
Abstract
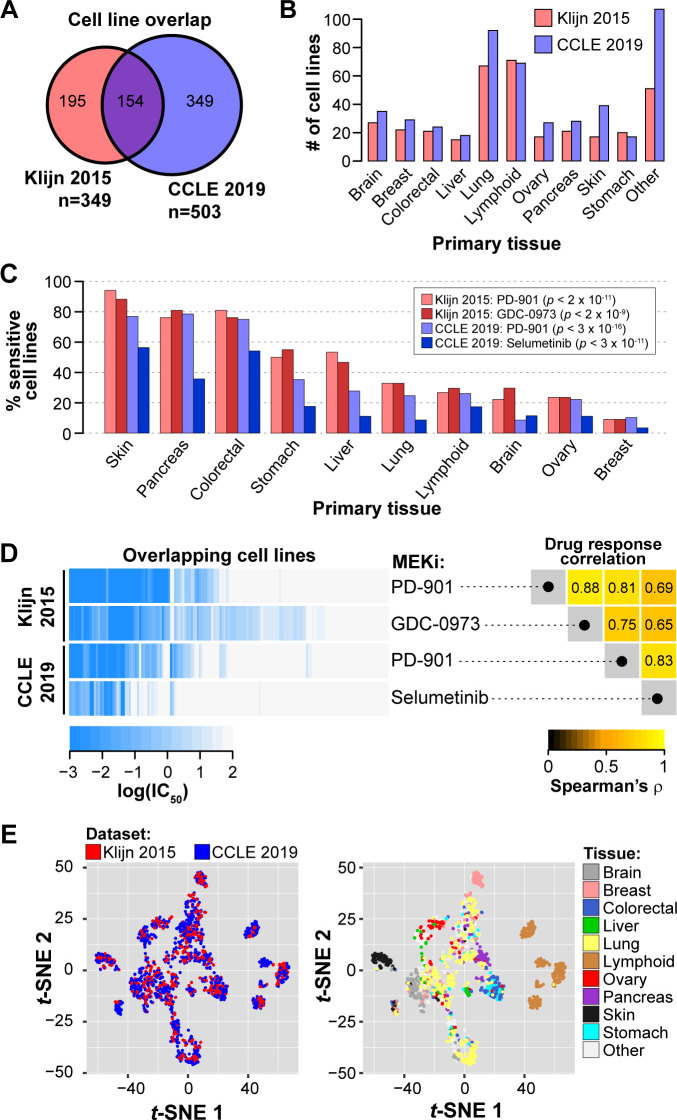
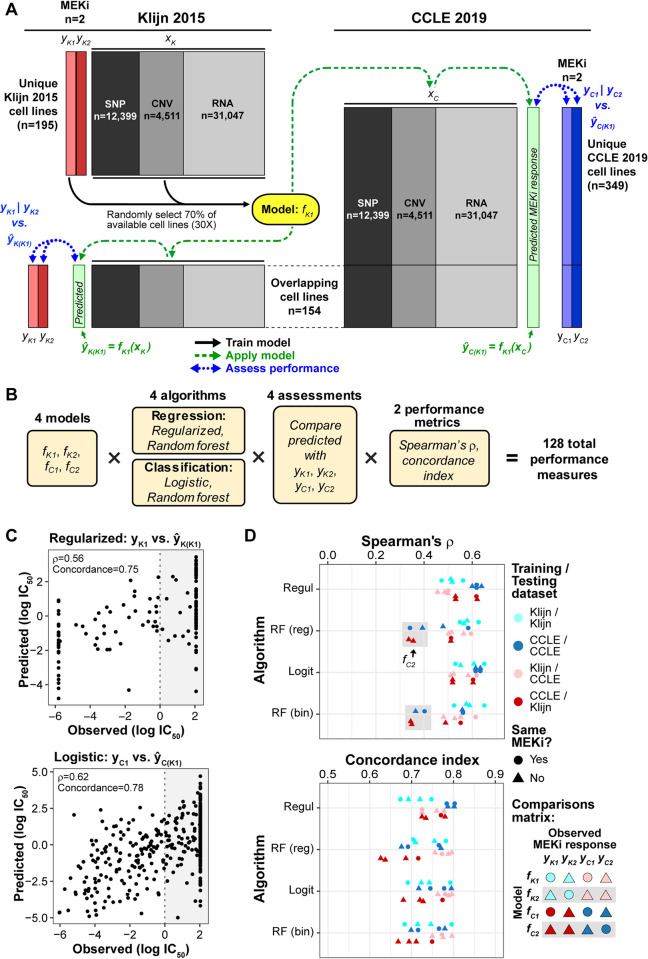
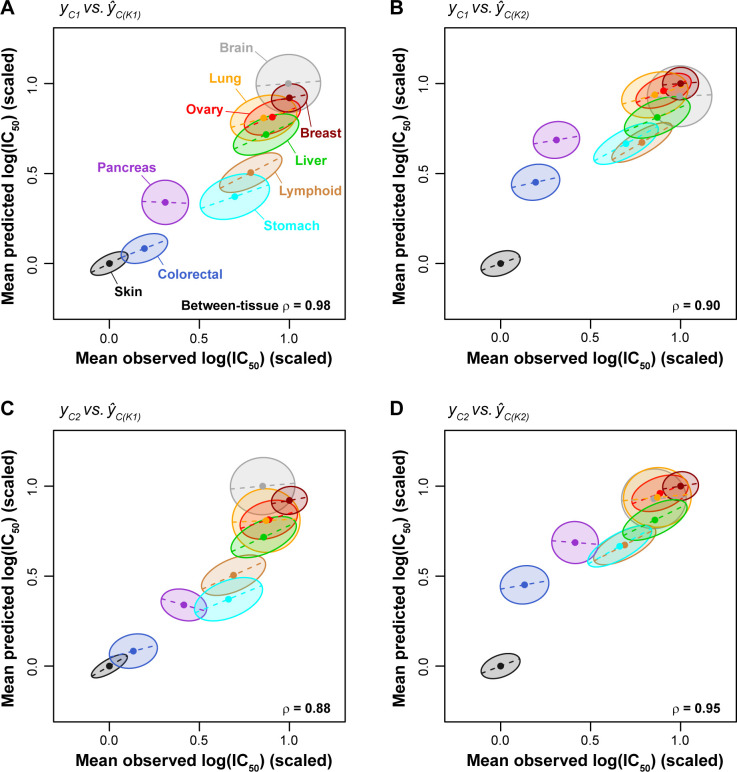
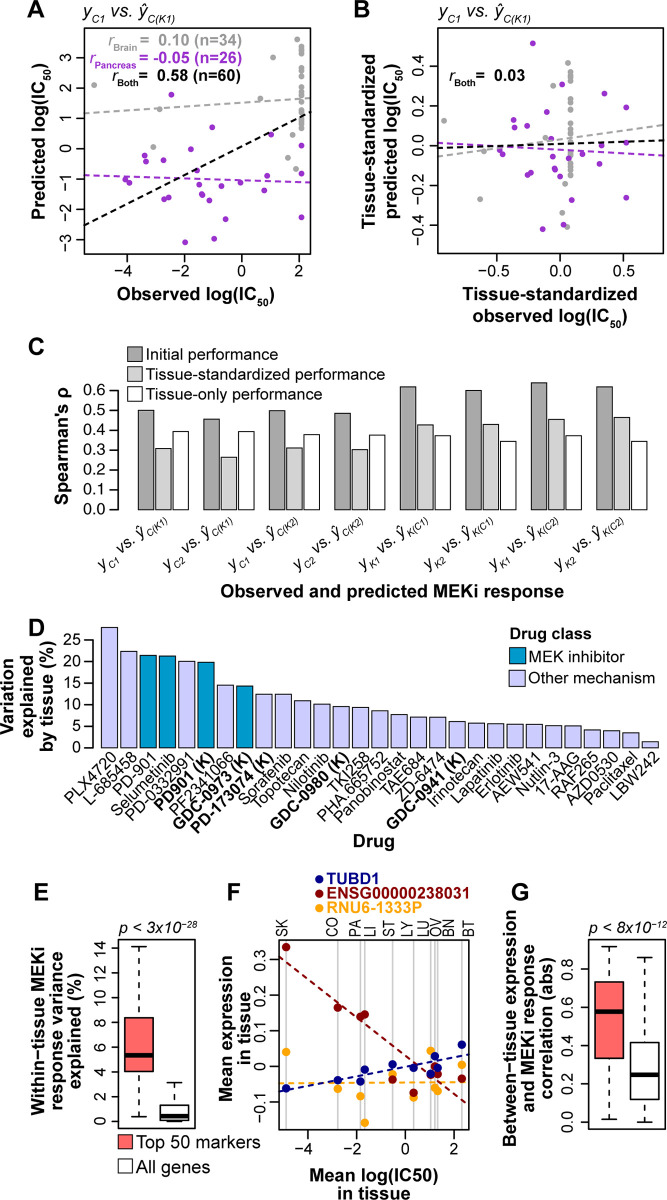
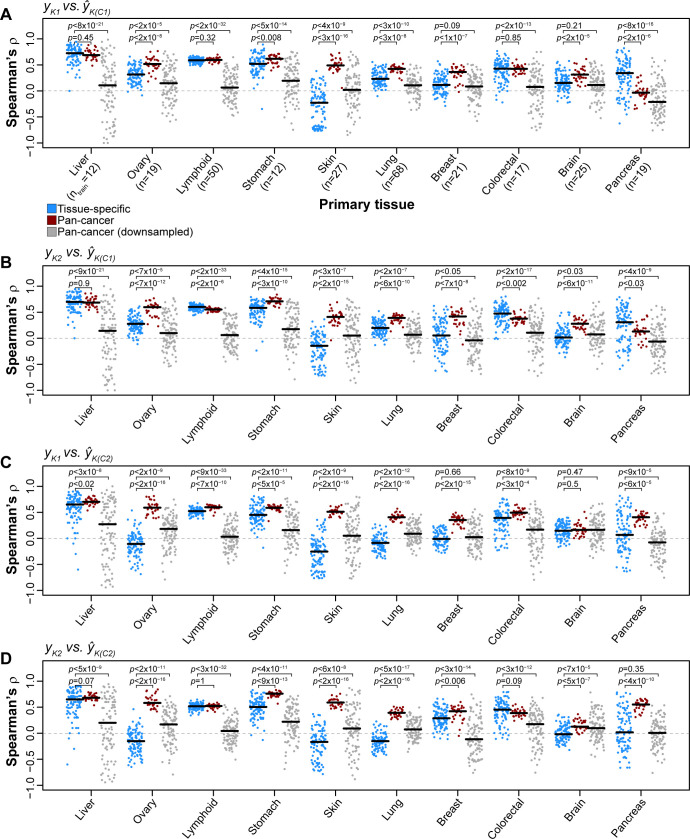
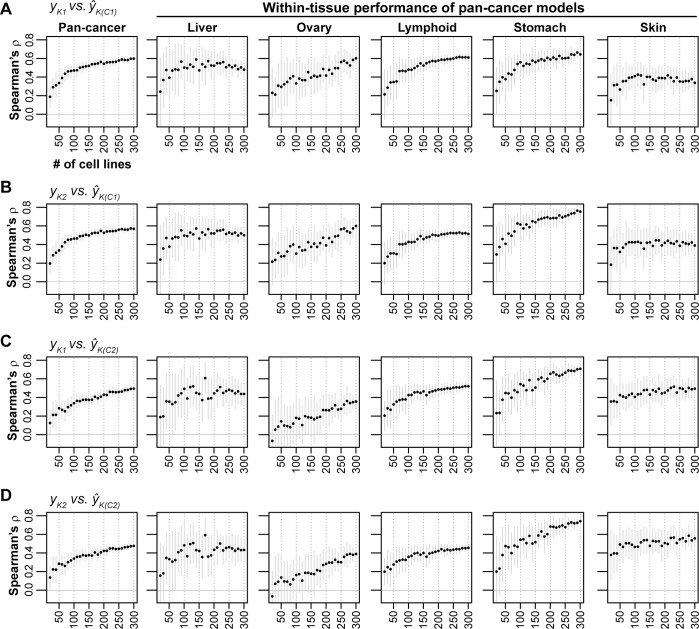
Increased availability of drug response and genomics data for many tumor cell lines has accelerated the development of pan-cancer prediction models of drug response. However, it is unclear how much between-tissue differences in drug response and molecular characteristics may contribute to pan-cancer predictions. Also unknown is whether the performance of pan-cancer models could vary by cancer type. Here, we built a series of pan-cancer models using two datasets containing 346 and 504 cell lines, each with MEK inhibitor (MEKi) response and mRNA expression, point mutation, and copy number variation data, and found that, while the tissue-level drug responses are accurately predicted (between-tissue ρ = 0.88-0.98), only 5 of 10 cancer types showed successful within-tissue prediction performance (within-tissue ρ = 0.11-0.64). Between-tissue differences make substantial contributions to the performance of pan-cancer MEKi response predictions, as exclusion of between-tissue signals leads to a decrease in Spearman's ρ from a range of 0.43-0.62 to 0.30-0.51. In practice, joint analysis of multiple cancer types usually has a larger sample size, hence greater power, than for one cancer type; and we observe that higher accuracy of pan-cancer prediction of MEKi response is almost entirely due to the sample size advantage. Success of pan-cancer prediction reveals how drug response in different cancers may invoke shared regulatory mechanisms despite tissue-specific routes of oncogenesis, yet predictions in different cancer types require flexible incorporation of between-cancer and within-cancer signals. As most datasets in genome sciences contain multiple levels of heterogeneity, careful parsing of group characteristics and within-group, individual variation is essential when making robust inference.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
