

Haplotype-resolved diverse human genomes and integrated analysis of structural variation
- PMID: 33632895
- PMCID: PMC8026704
- DOI: 10.1126/science.abf7117
Haplotype-resolved diverse human genomes and integrated analysis of structural variation
Abstract
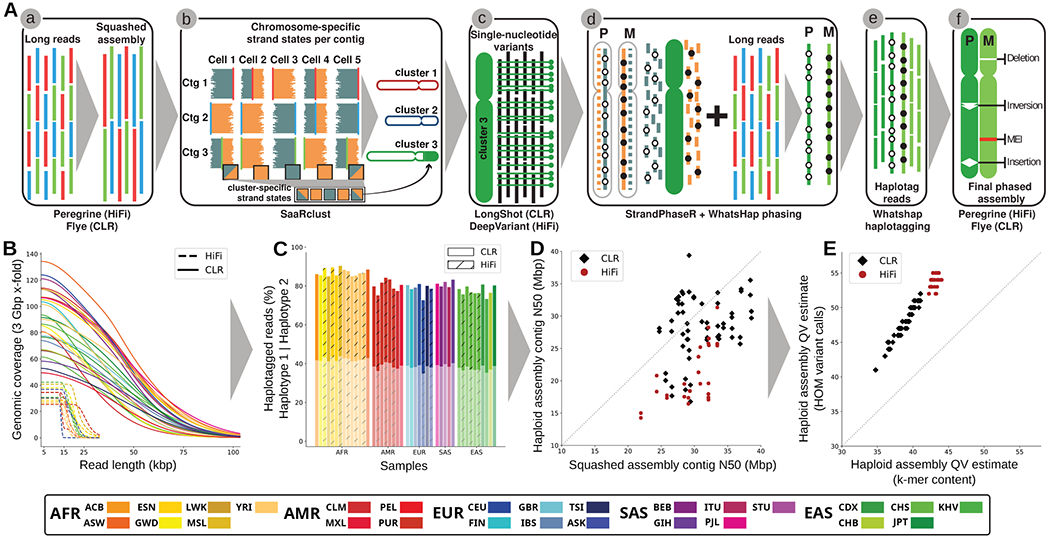
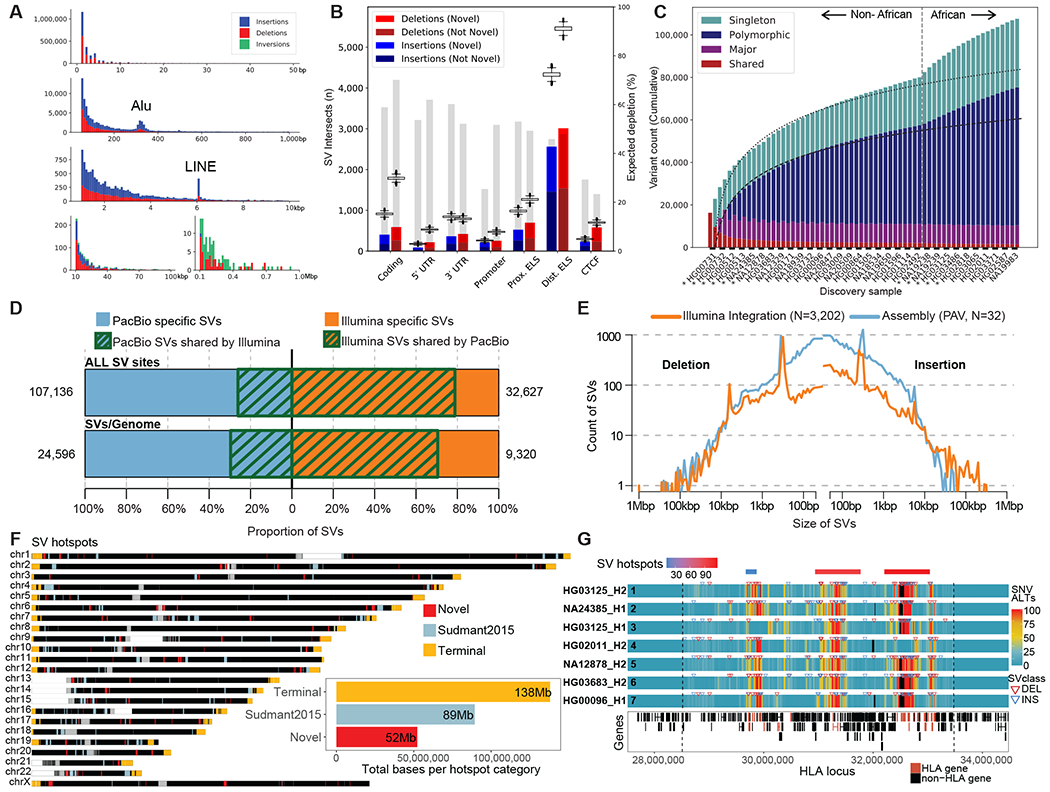
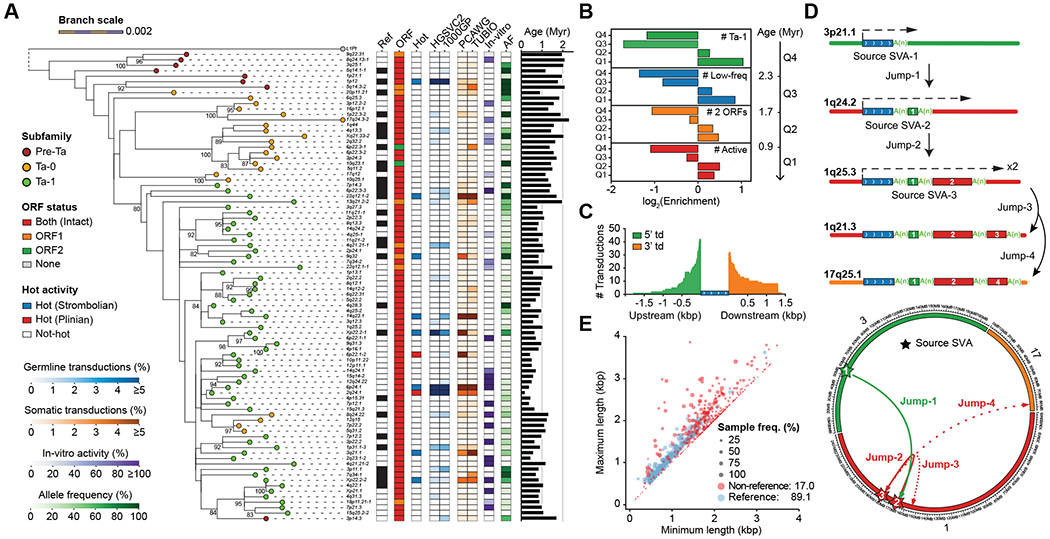
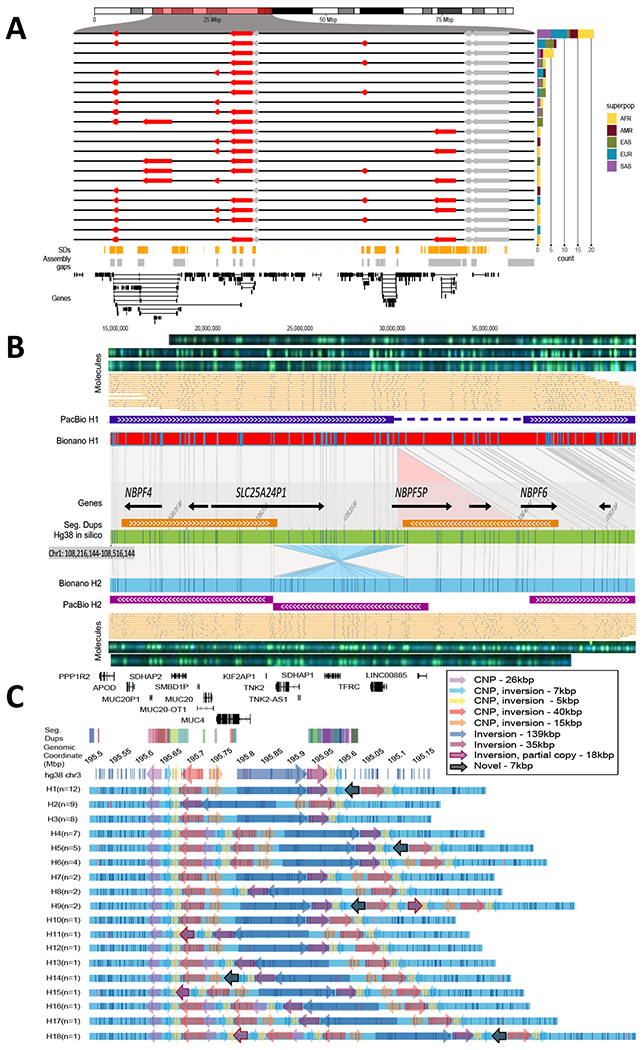
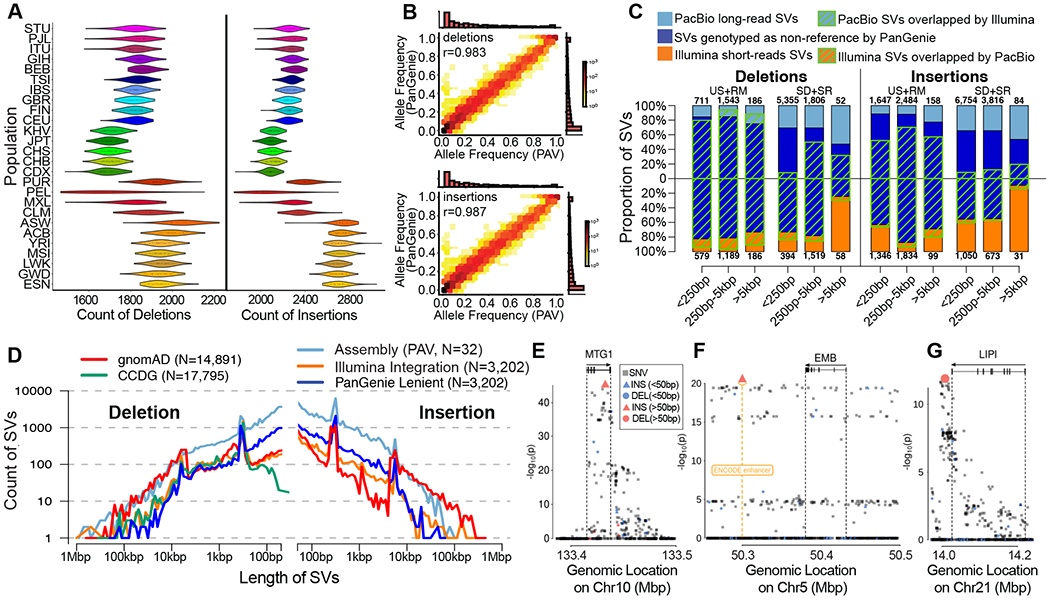
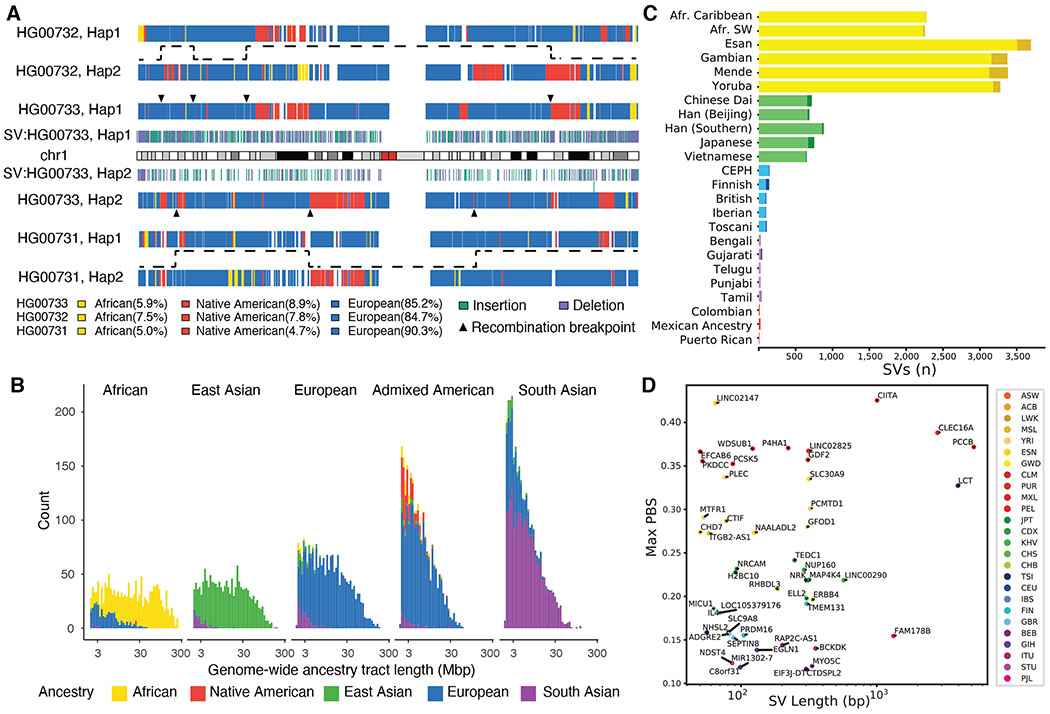
Long-read and strand-specific sequencing technologies together facilitate the de novo assembly of high-quality haplotype-resolved human genomes without parent-child trio data. We present 64 assembled haplotypes from 32 diverse human genomes. These highly contiguous haplotype assemblies (average minimum contig length needed to cover 50% of the genome: 26 million base pairs) integrate all forms of genetic variation, even across complex loci. We identified 107,590 structural variants (SVs), of which 68% were not discovered with short-read sequencing, and 278 SV hotspots (spanning megabases of gene-rich sequence). We characterized 130 of the most active mobile element source elements and found that 63% of all SVs arise through homology-mediated mechanisms. This resource enables reliable graph-based genotyping from short reads of up to 50,340 SVs, resulting in the identification of 1526 expression quantitative trait loci as well as SV candidates for adaptive selection within the human population.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
Genome-wide analysis of structural variation.Nat Methods. 2021 May;18(5):448. doi: 10.1038/s41592-021-01161-z. Nat Methods. 2021. PMID: 33963350 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R35 GM138212/GM/NIGMS NIH HHS/United States
- U01 HG010973/HG/NHGRI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- 773026/ERC_/European Research Council/International
- R01 HD081256/HD/NICHD NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- U24 HG007497/HG/NHGRI NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- R15 HG009565/HG/NHGRI NIH HHS/United States
- T32 DK067872/DK/NIDDK NIH HHS/United States
- R01 HG002898/HG/NHGRI NIH HHS/United States
- K99 HG011041/HG/NHGRI NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- R01 HG007068/HG/NHGRI NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- OT3 HL147154/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
