

Lentivirus-mediated gene therapy for Fabry disease
- PMID: 33633114
- PMCID: PMC7907075
- DOI: 10.1038/s41467-021-21371-5
Lentivirus-mediated gene therapy for Fabry disease
Abstract
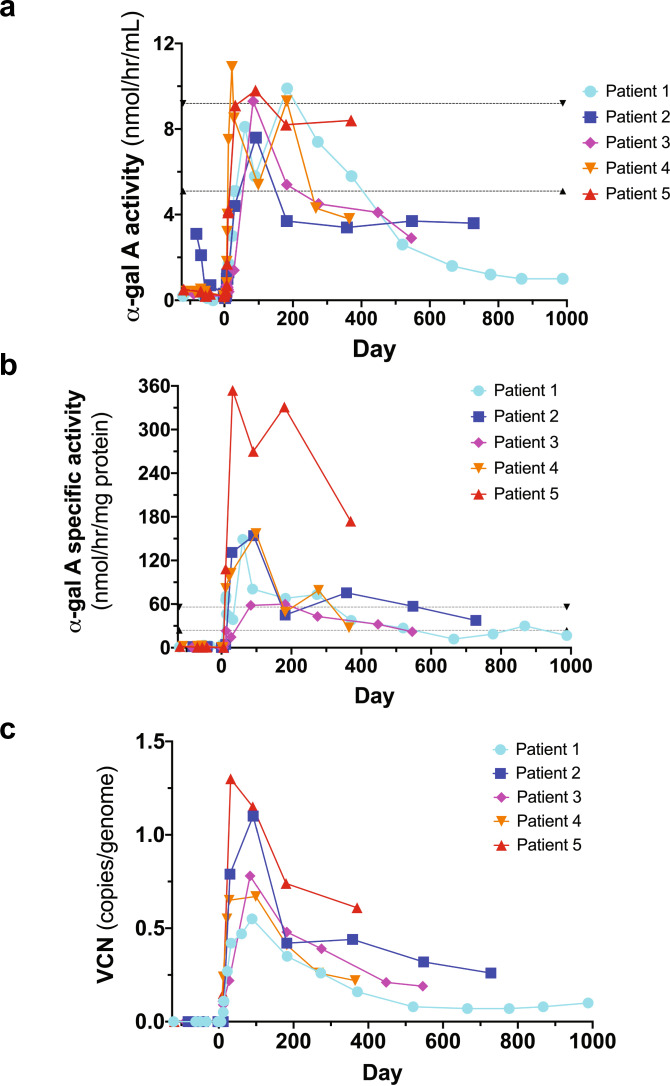
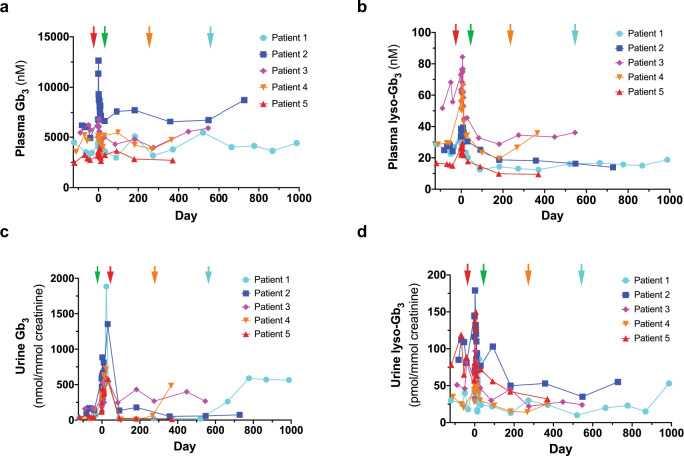
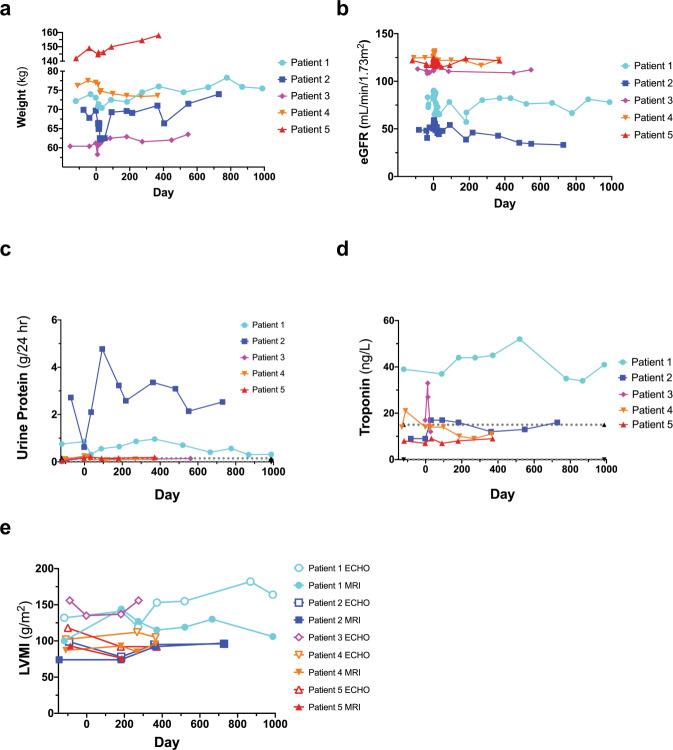
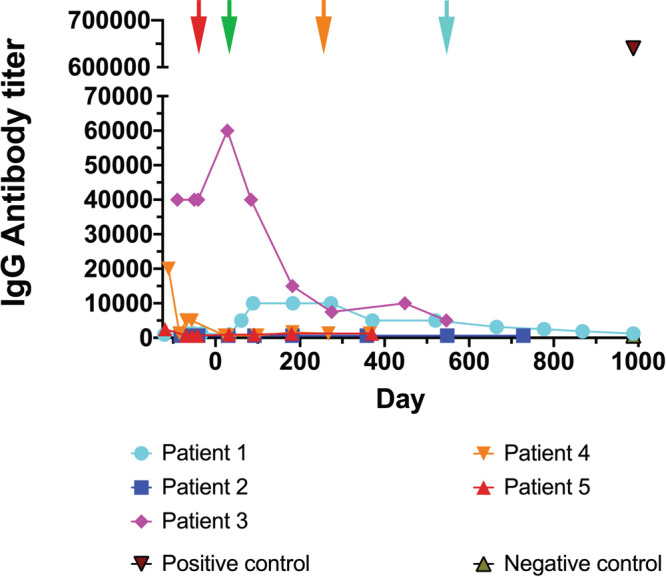
Enzyme and chaperone therapies are used to treat Fabry disease. Such treatments are expensive and require intrusive biweekly infusions; they are also not particularly efficacious. In this pilot, single-arm study (NCT02800070), five adult males with Type 1 (classical) phenotype Fabry disease were infused with autologous lentivirus-transduced, CD34+-selected, hematopoietic stem/progenitor cells engineered to express alpha-galactosidase A (α-gal A). Safety and toxicity are the primary endpoints. The non-myeloablative preparative regimen consisted of intravenous melphalan. No serious adverse events (AEs) are attributable to the investigational product. All patients produced α-gal A to near normal levels within one week. Vector is detected in peripheral blood and bone marrow cells, plasma and leukocytes demonstrate α-gal A activity within or above the reference range, and reductions in plasma and urine globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) are seen. While the study and evaluations are still ongoing, the first patient is nearly three years post-infusion. Three patients have elected to discontinue enzyme therapy.
Conflict of interest statement
A.K. received grants, consulting fees, revenue distribution agreement, speaker fees and travel support with AVROBIO, Inc. as well as revenue distribution agreement with University Health Network regarding gene therapy using technology from this work. D.L.B. and J.H. were partially paid from a Sponsored Research Agreement—AVROBIO, Inc. C. A. Rupar has the following financial relationships to disclose: the Biochemical Genetics clinical diagnostic laboratory at his home institution is contracted by AVROBIO, Inc. to assay enzymes on a fee for service basis. He is the laboratory director but receives no personal compensation. C.A.-B. has received a service contract and honoraria for biomarker analysis with AVROBIO, Inc., grant from CIHR. K.G. had travel paid for by AVROBIO, Inc. S.W. has received nonfinancial support from Sanofi-Genzyme, nonfinancial support from Takeda Pharmaceuticals (formerly Shire HGT), personal fees and nonfinancial support from Amicus Therapeutics. K.L. has received travel grant and honorarium from Amicus Therapies; travel grant and speaker fees from Sanofi-Genzyme; travel grant, consulting fees and speaker fees from Takeda Pharmaceuticals; medical advisor to the Canadian Fabry Disease Association. C.F.M. has received grants, personal fees, and nonfinancial support from Takeda Pharmaceuticals (previously Shire HGT), grants, personal fees and nonfinancial support from Sanofi-Genzyme, nonfinancial support from Amicus Therapeutics. A.K. has received consultancy fees from AVROBIO, Inc. unrelated to this study. M.L.W. has received research grants, consulting fees, speaker fees and travel support with Amicus Therapeutics, Protalix, Sanofi-Genzyme and Takeda, revenue distribution agreement with University Health Network regarding gene therapy using technology from this work. J.A.M. has the following financial relationships to disclose: SAB—Rapa Therapeutics. Honoraria—Sanofi-Genzyme, Shire. Co-Founder—AVROBIO, Inc. Shareholder—AVROBIO, Inc. Grants from Canadian Institutes of Health Research and Kidney Foundation of Canada and AVROBIO, Inc. M.B., A.C., P.R.D., R.F., D.H.F., G.F., S.J., W.M.M., P.O., N.P., and J.W.R. have no financial relationships to disclose in relation to this trial.
Figures
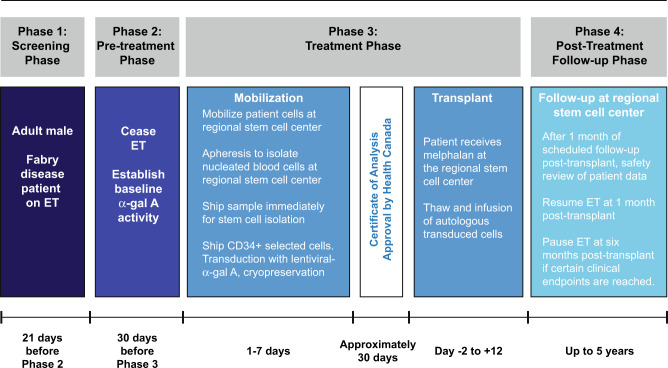
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
