

ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis
- PMID: 33633365
- PMCID: PMC8012210
- DOI: 10.1038/s41588-021-00790-6
ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis
Erratum in
-
Author Correction: ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis.Nat Genet. 2021 Jun;53(6):935. doi: 10.1038/s41588-021-00850-x. Nat Genet. 2021. PMID: 33790476 Free PMC article. No abstract available.
Abstract
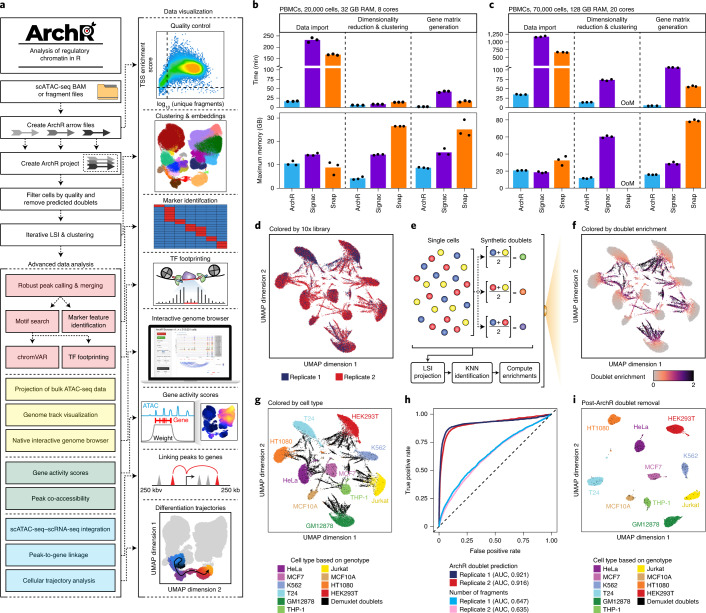
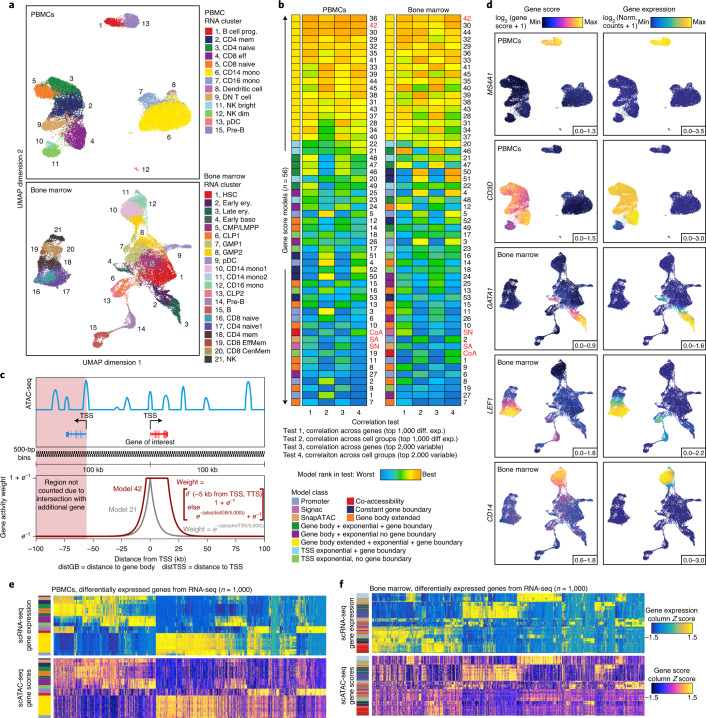
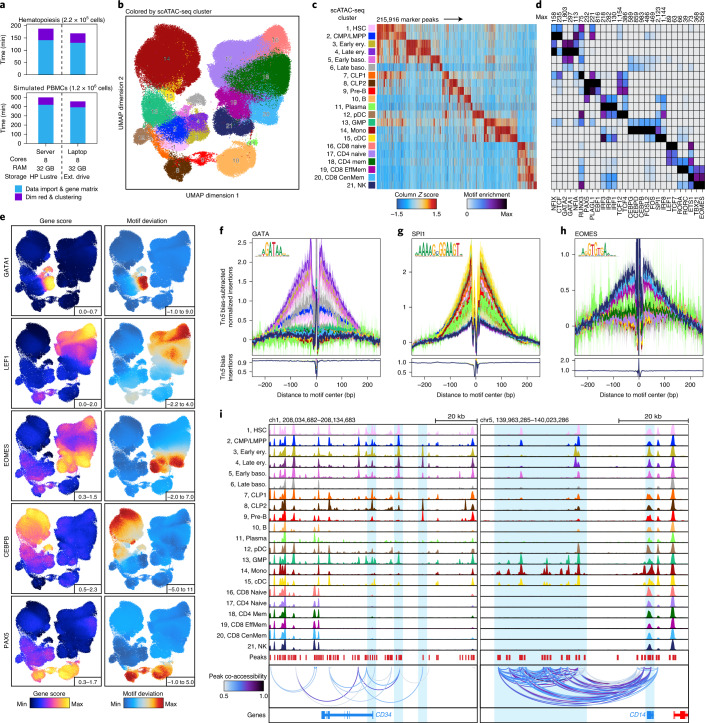
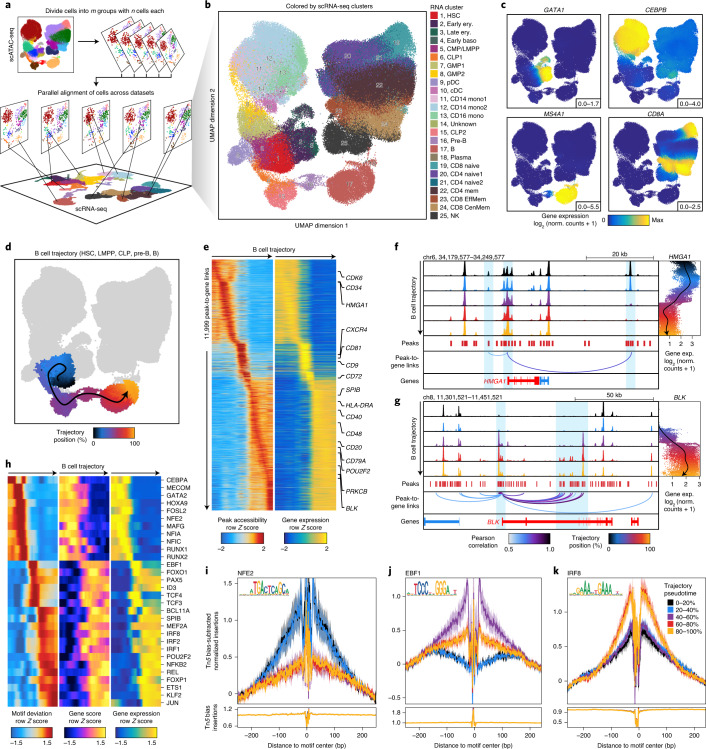
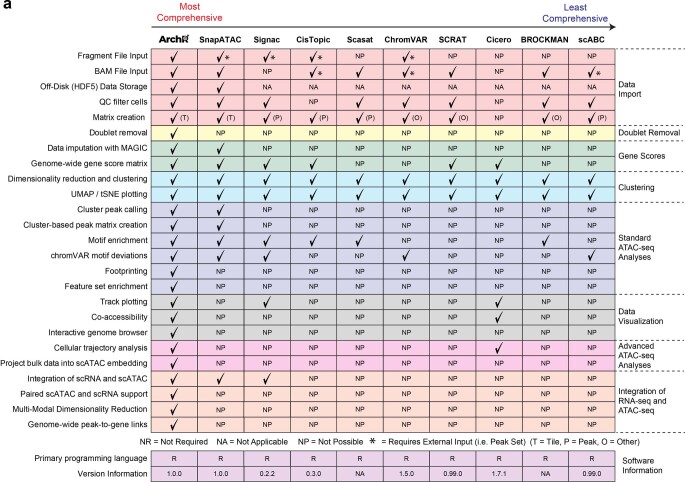
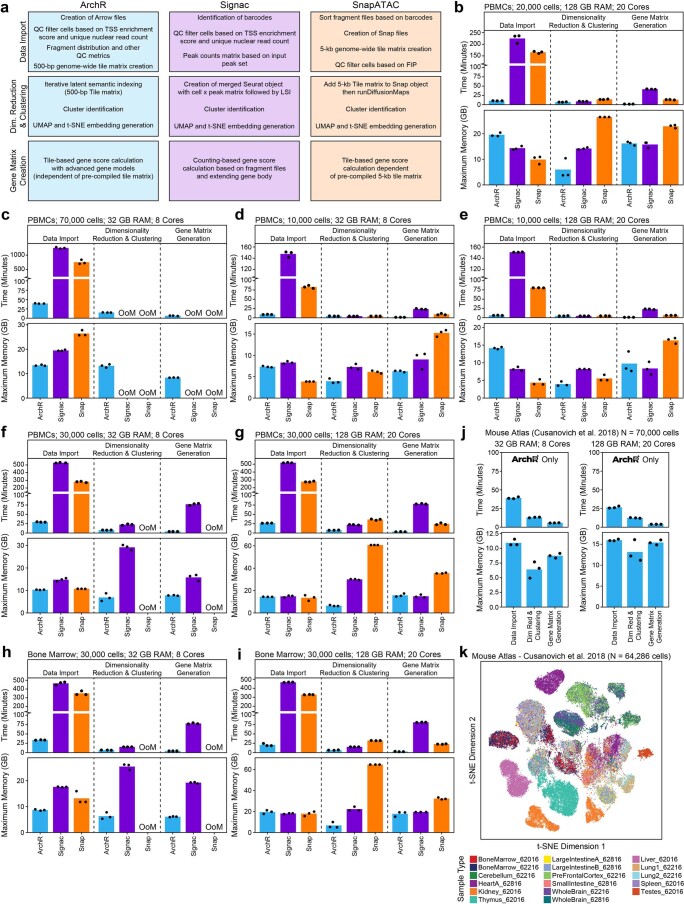
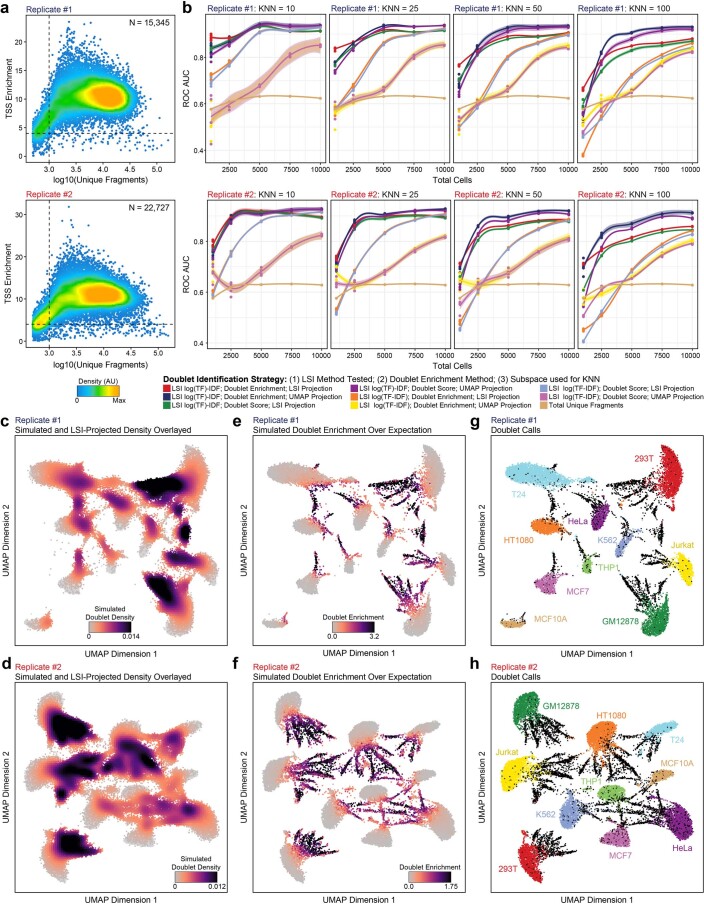
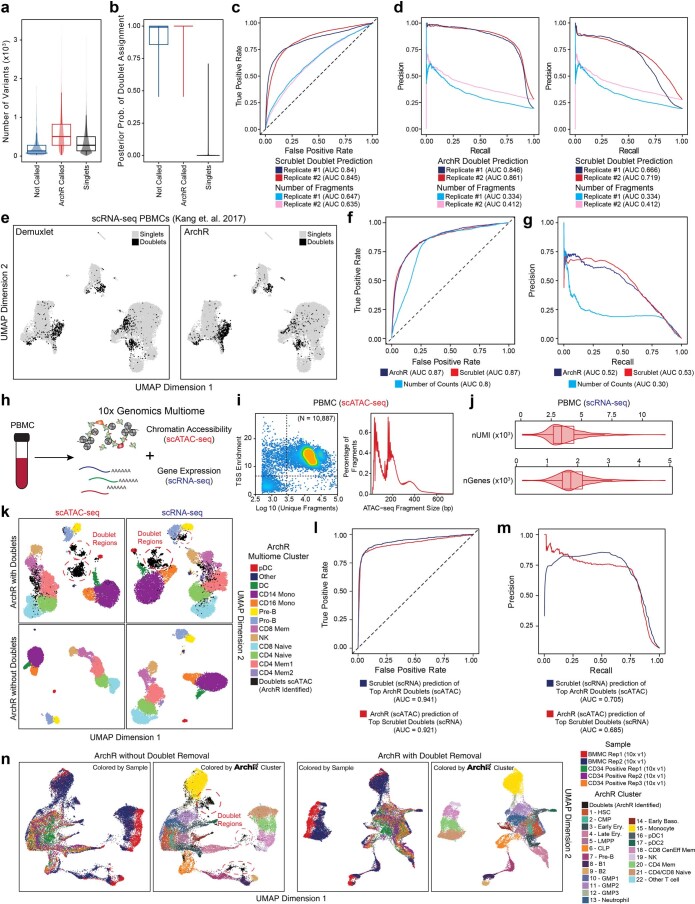
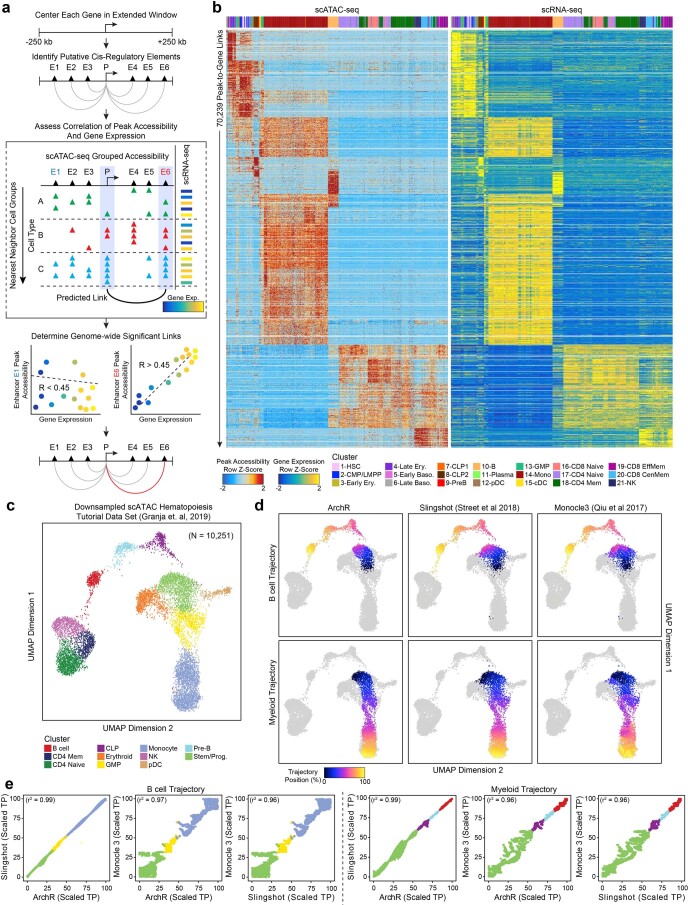
The advent of single-cell chromatin accessibility profiling has accelerated the ability to map gene regulatory landscapes but has outpaced the development of scalable software to rapidly extract biological meaning from these data. Here we present a software suite for single-cell analysis of regulatory chromatin in R (ArchR; https://www.archrproject.com/ ) that enables fast and comprehensive analysis of single-cell chromatin accessibility data. ArchR provides an intuitive, user-focused interface for complex single-cell analyses, including doublet removal, single-cell clustering and cell type identification, unified peak set generation, cellular trajectory identification, DNA element-to-gene linkage, transcription factor footprinting, mRNA expression level prediction from chromatin accessibility and multi-omic integration with single-cell RNA sequencing (scRNA-seq). Enabling the analysis of over 1.2 million single cells within 8 h on a standard Unix laptop, ArchR is a comprehensive software suite for end-to-end analysis of single-cell chromatin accessibility that will accelerate the understanding of gene regulation at the resolution of individual cells.
Conflict of interest statement
W.J.G. and H.Y.C. are consultants for 10x Genomics, which has licensed IP associated with ATAC-seq. W.J.G. has additional affiliations with Guardant Health (consultant) and Protillion Biosciences (cofounder and consultant). H.Y.C. is a cofounder of Accent Therapeutics and Boundless Bio and is a consultant for Arsenal Biosciences and Spring Discovery.
Figures
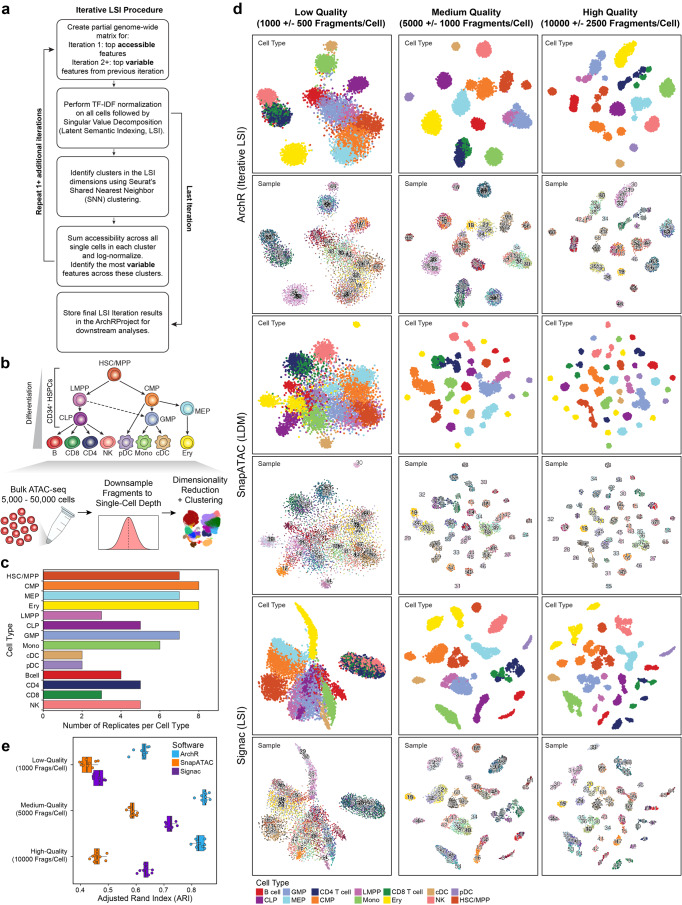
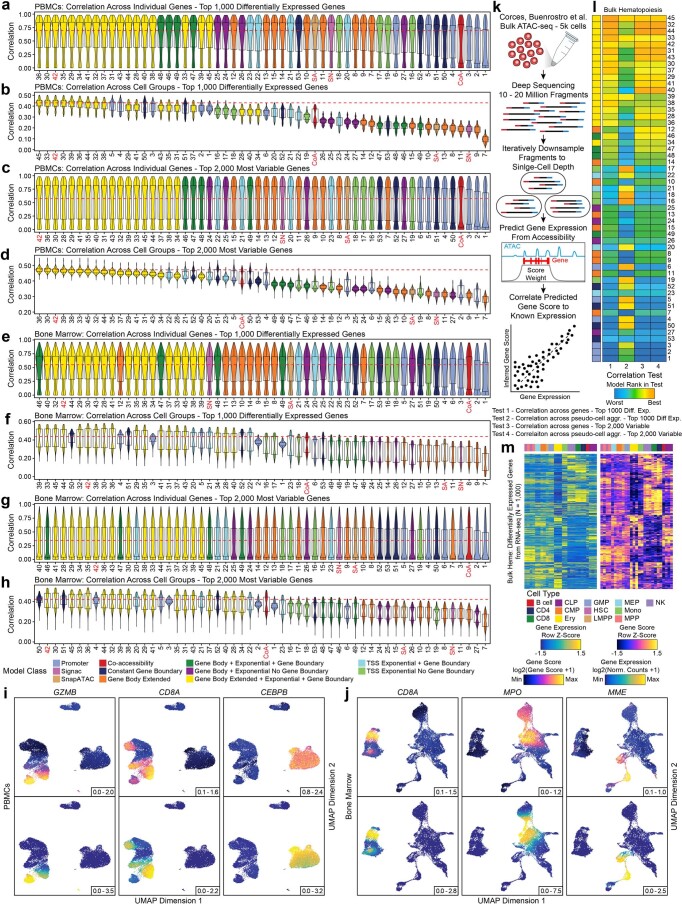
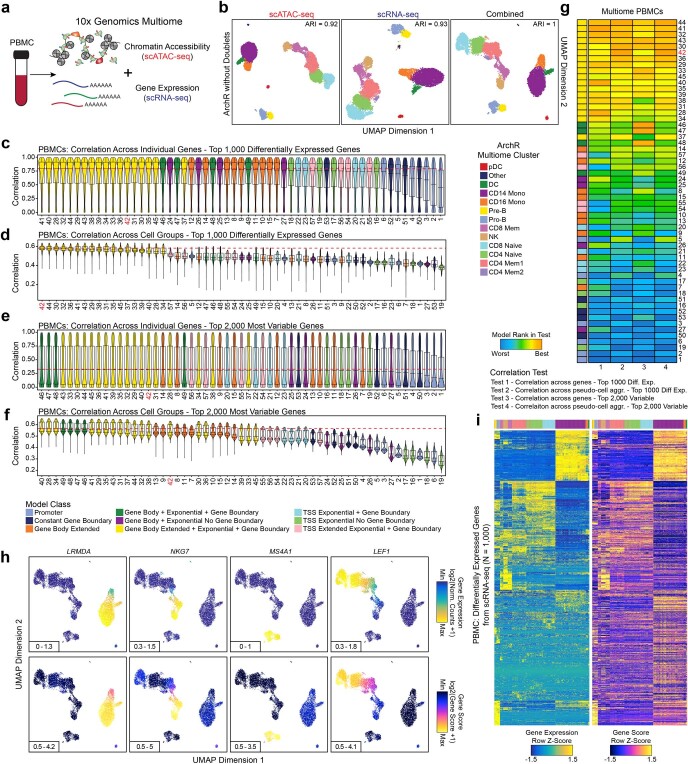
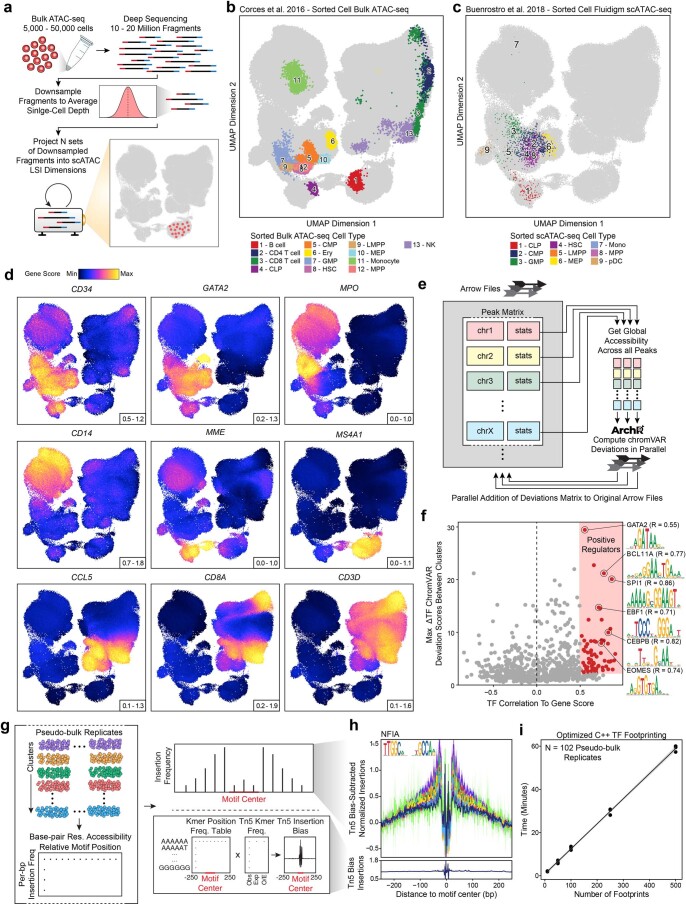

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
