

An emerging role for cellular crosstalk in the cancer stem cell niche
- PMID: 33634866
- PMCID: PMC9575701
- DOI: 10.1002/path.5655
An emerging role for cellular crosstalk in the cancer stem cell niche
Abstract
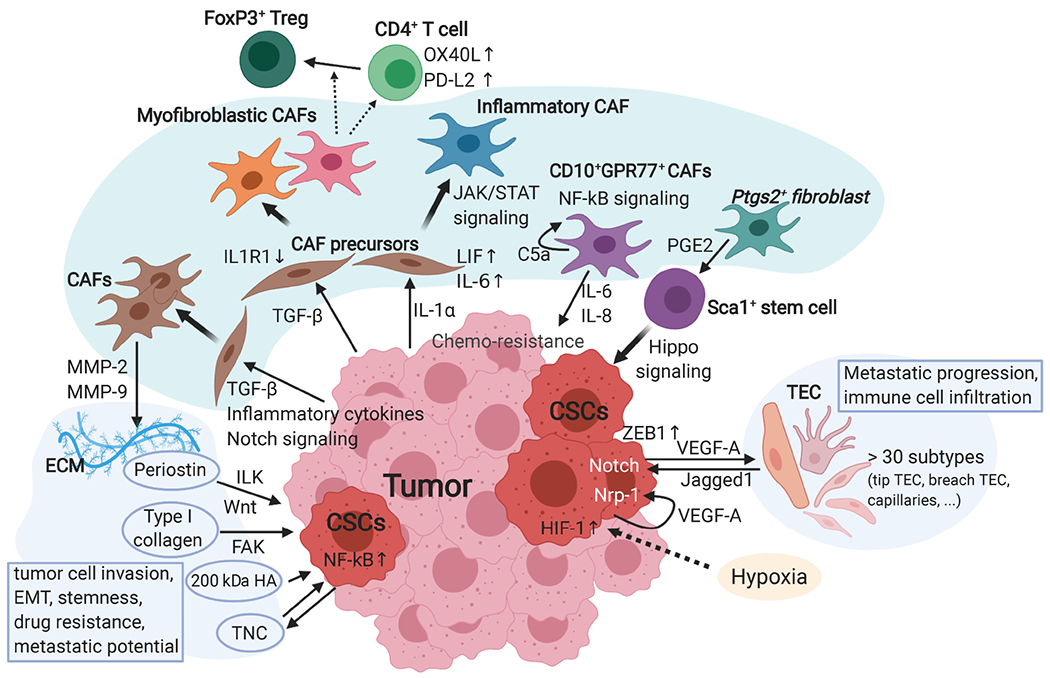
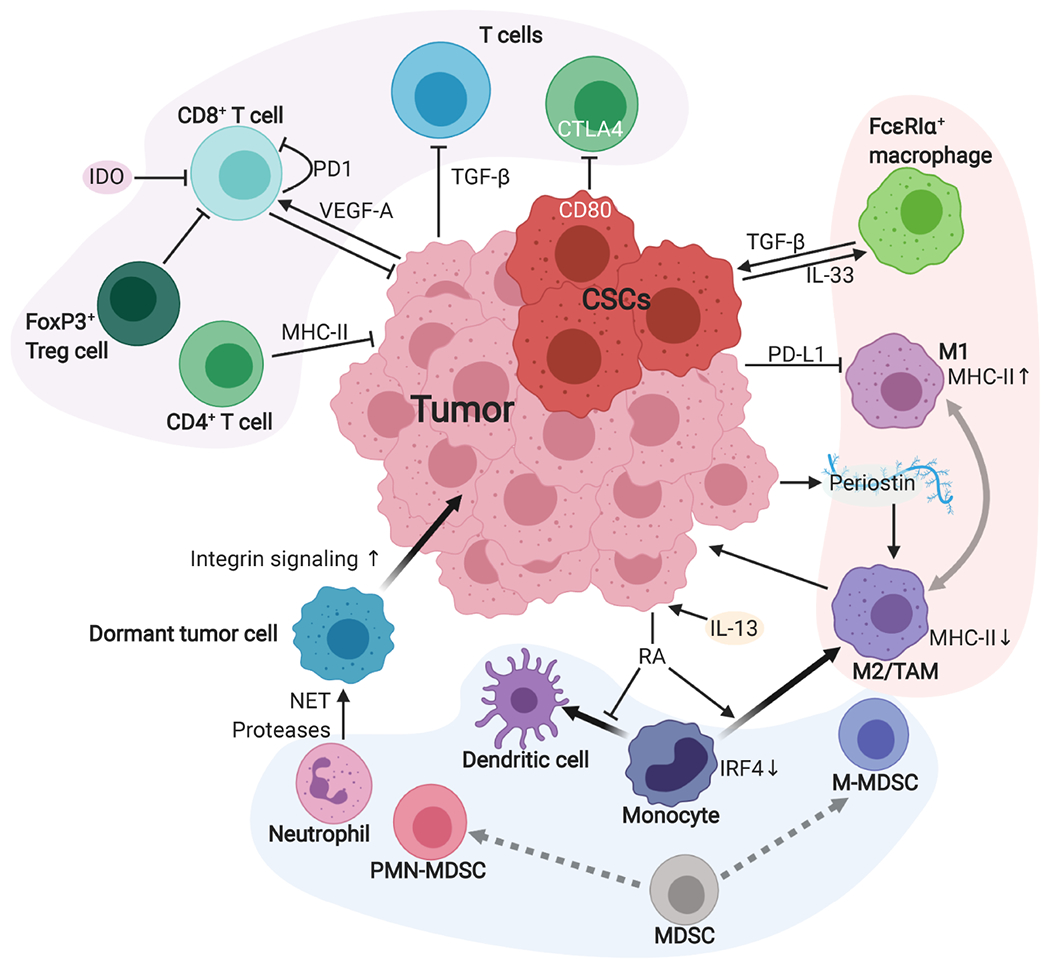
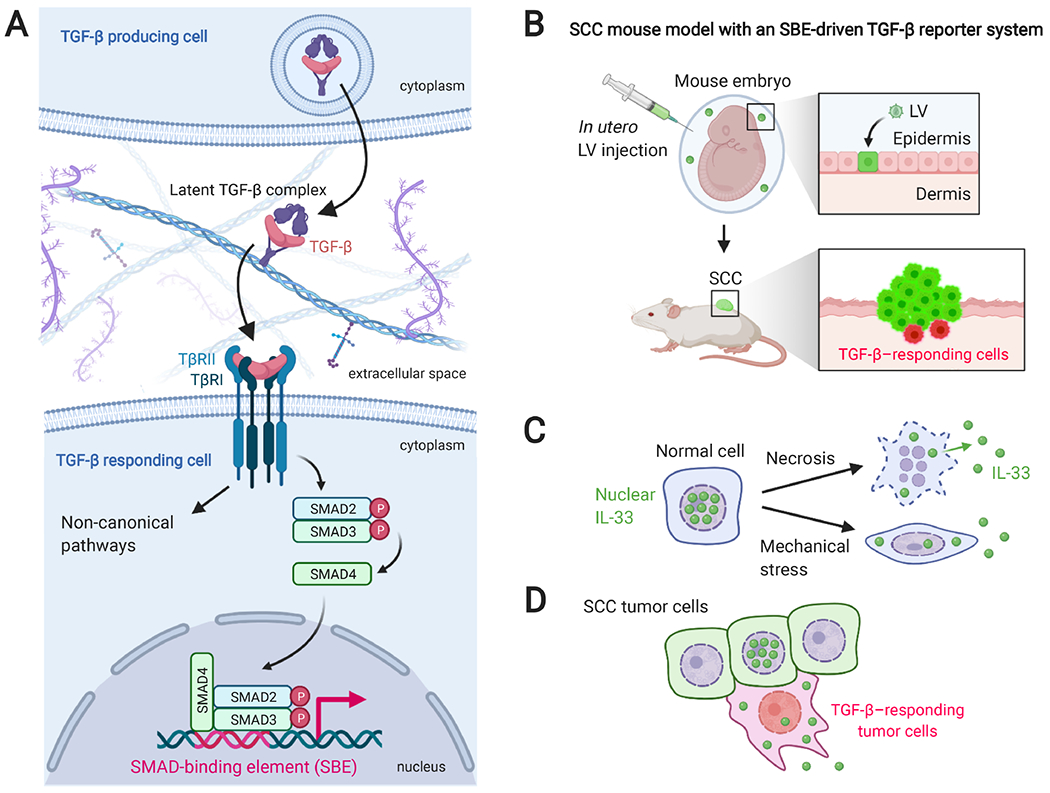
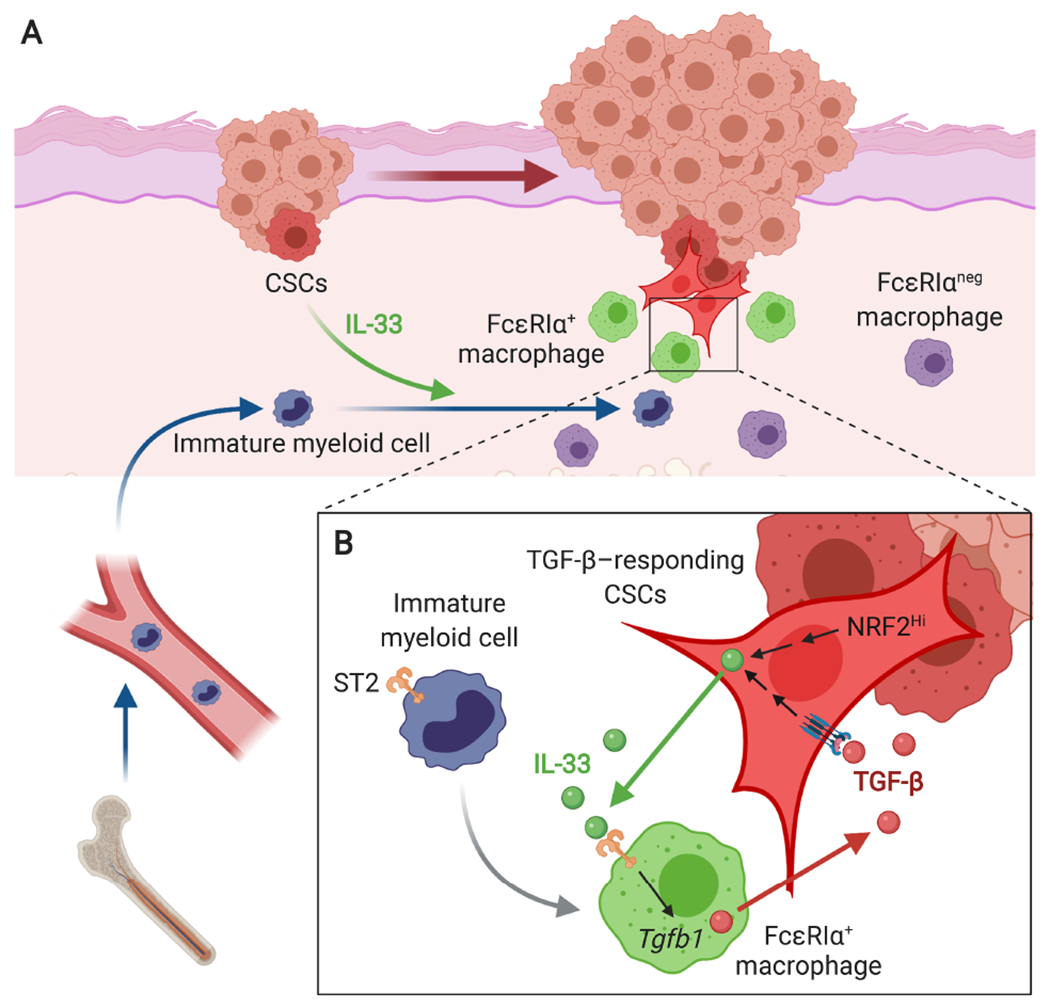
Although cumulative genetic and epigenetic changes in cancer cells are correlated with tumor malignancy, accumulating evidence supports that tumor cell-extrinsic mechanisms play an essential role in driving tumor progression. The tissue architecture surrounding tumor cells evolves during disease progression and becomes a significant barrier to cancer treatments. The functional traits of the tumor microenvironment (TME), either tumor suppressive or supportive, are defined by the distribution of various stromal cells and their sequential and reciprocal cellular interactions. Recent studies have uncovered a significant heterogeneity in stromal cells and identified specific subpopulations correlated with clinical outcomes, providing novel insights into the complex TME system that drives tumor progression and therapy resistance. Moreover, a small population of tumor cells with tumor-initiating and drug-resistant capabilities, cancer stem cells (CSCs), is maintained by the specialized TME, the so-called CSC niche. The crosstalk between CSCs and niche cells is an attractive avenue for identifying the vulnerability of difficult-to-treat cancers. Here, we review the recent advance in understanding TME biology and its impact on CSCs. We then focus on a newly identified niche signaling loop by which CSCs promote malignant progression and drug resistance of squamous cell carcinoma. The CSC niche is a promising research field that needs more attention and could facilitate the development of durable cancer treatment. © 2021 The Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: CSC niche; IL-33; TGF-β; cancer progression; cancer stem cells (CSCs); cellular crosstalk; drug resistance; macrophages; tumor microenvironment (TME).
© 2021 The Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Conflict of interest statement
No conflicts of interest were declared.
Figures
Similar articles
-
Cancer stem cells and their niche in the progression of squamous cell carcinoma.Cancer Sci. 2020 Nov;111(11):3985-3992. doi: 10.1111/cas.14639. Epub 2020 Sep 18. Cancer Sci. 2020. PMID: 32888236 Free PMC article. Review.
-
Cancer stem cells and tumor-associated macrophages as mates in tumor progression: mechanisms of crosstalk and advanced bioinformatic tools to dissect their phenotypes and interaction.Front Immunol. 2025 Feb 6;16:1529847. doi: 10.3389/fimmu.2025.1529847. eCollection 2025. Front Immunol. 2025. PMID: 39981232 Free PMC article. Review.
-
Cancer stem cells (CSCs) in cancer progression and therapy.J Cell Physiol. 2019 Jun;234(6):8381-8395. doi: 10.1002/jcp.27740. Epub 2018 Nov 11. J Cell Physiol. 2019. PMID: 30417375 Review.
-
Exosome crosstalk between cancer stem cells and tumor microenvironment: cancer progression and therapeutic strategies.Stem Cell Res Ther. 2024 Nov 22;15(1):449. doi: 10.1186/s13287-024-04061-z. Stem Cell Res Ther. 2024. PMID: 39578849 Free PMC article. Review.
-
Exploring the dynamic interplay between cancer stem cells and the tumor microenvironment: implications for novel therapeutic strategies.J Transl Med. 2023 Oct 2;21(1):686. doi: 10.1186/s12967-023-04575-9. J Transl Med. 2023. PMID: 37784157 Free PMC article. Review.
Cited by
-
A Gold Nanoparticle Bioconjugate Delivery System for Active Targeted Photodynamic Therapy of Cancer and Cancer Stem Cells.Cancers (Basel). 2022 Sep 20;14(19):4558. doi: 10.3390/cancers14194558. Cancers (Basel). 2022. PMID: 36230480 Free PMC article. Review.
-
Evaluation of regulatory T-cells in cancer immunotherapy: therapeutic relevance of immune checkpoint inhibition.Med Oncol. 2024 Jan 18;41(2):59. doi: 10.1007/s12032-023-02289-y. Med Oncol. 2024. PMID: 38238513 Review.
-
Lymph Vessels Associate with Cancer Stem Cells from Initiation to Malignant Stages of Squamous Cell Carcinoma.Int J Mol Sci. 2023 Sep 2;24(17):13615. doi: 10.3390/ijms241713615. Int J Mol Sci. 2023. PMID: 37686421 Free PMC article.
-
Cancer stem cells and their niche in cancer progression and therapy.Cancer Cell Int. 2023 Dec 1;23(1):305. doi: 10.1186/s12935-023-03130-2. Cancer Cell Int. 2023. PMID: 38041196 Free PMC article. Review.
-
Cancer stem cells: landscape, challenges and emerging therapeutic innovations.Signal Transduct Target Ther. 2025 Aug 5;10(1):248. doi: 10.1038/s41392-025-02360-2. Signal Transduct Target Ther. 2025. PMID: 40759634 Free PMC article. Review.
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. - PubMed
-
- Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov 2017; 16: 241–263. - PubMed
-
- Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013; 501: 346–354. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
