

Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells
- PMID: 33635334
- PMCID: PMC7948300
- DOI: 10.1182/bloodadvances.2020001996
Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells
Abstract
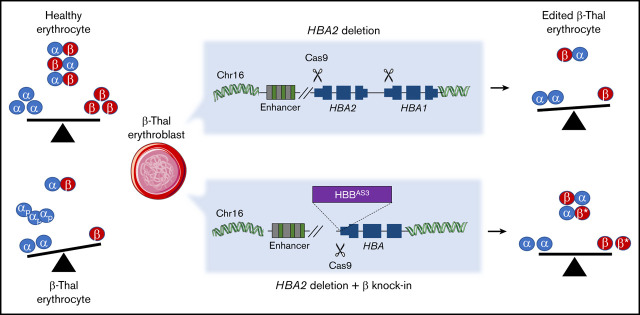
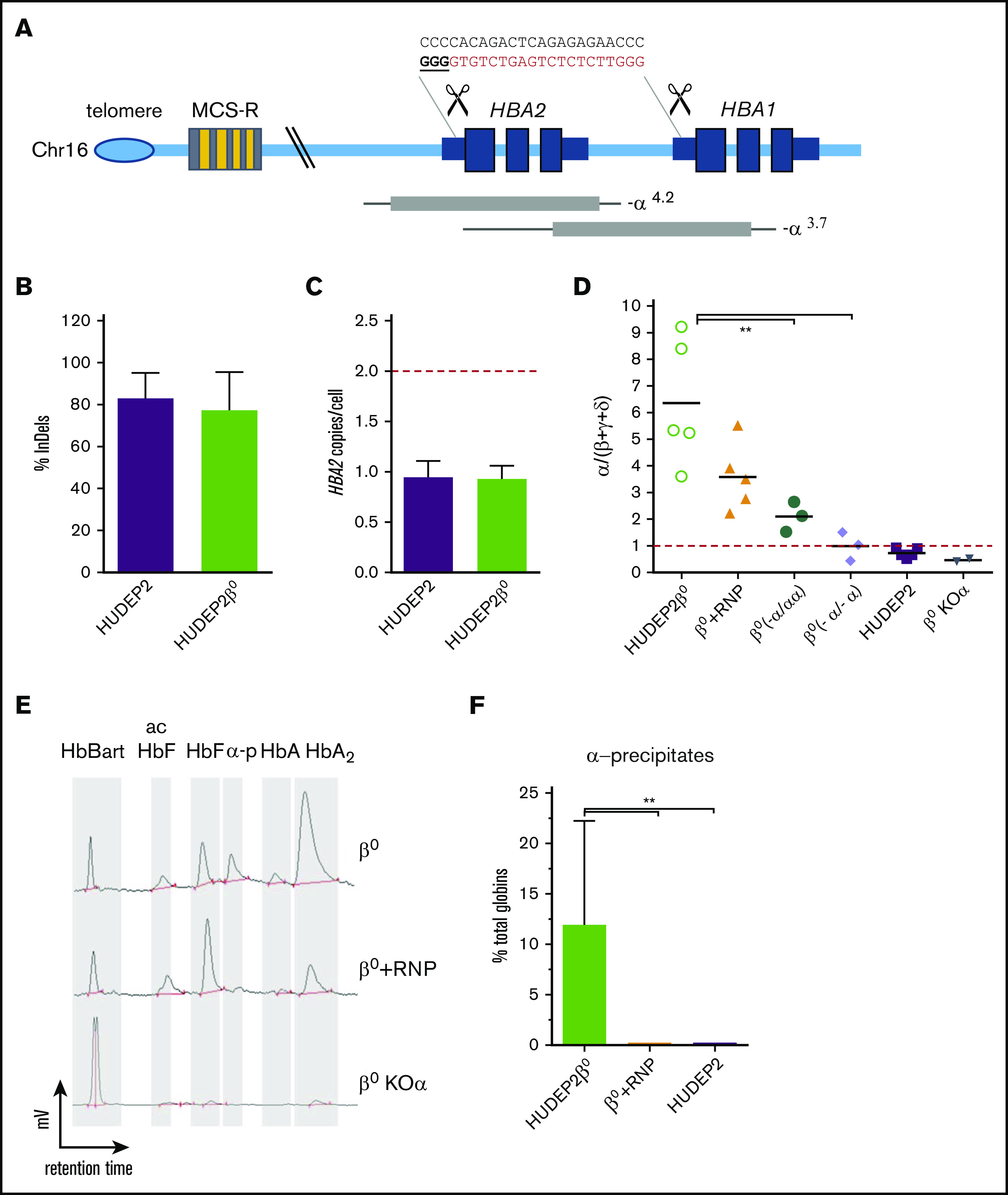
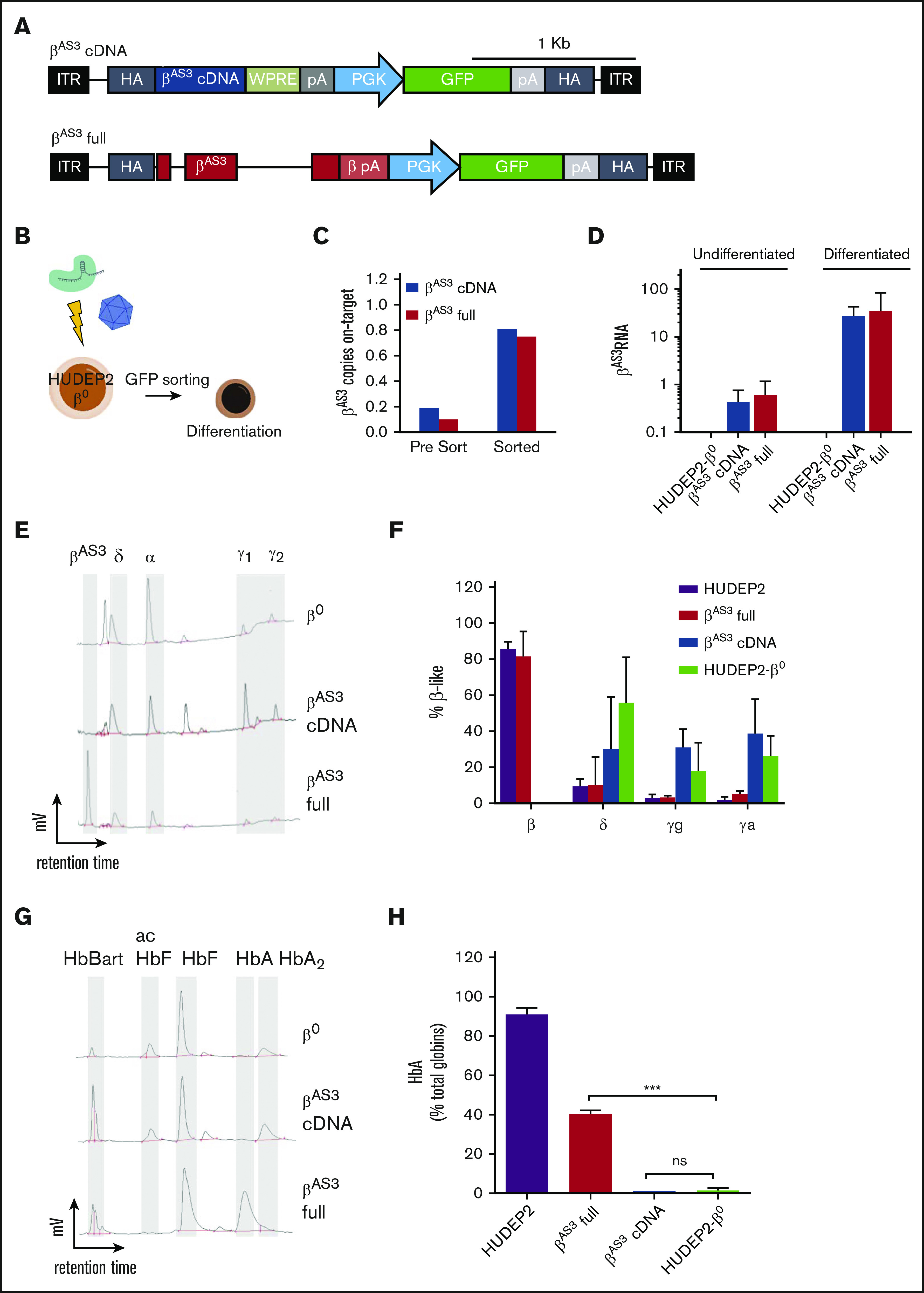
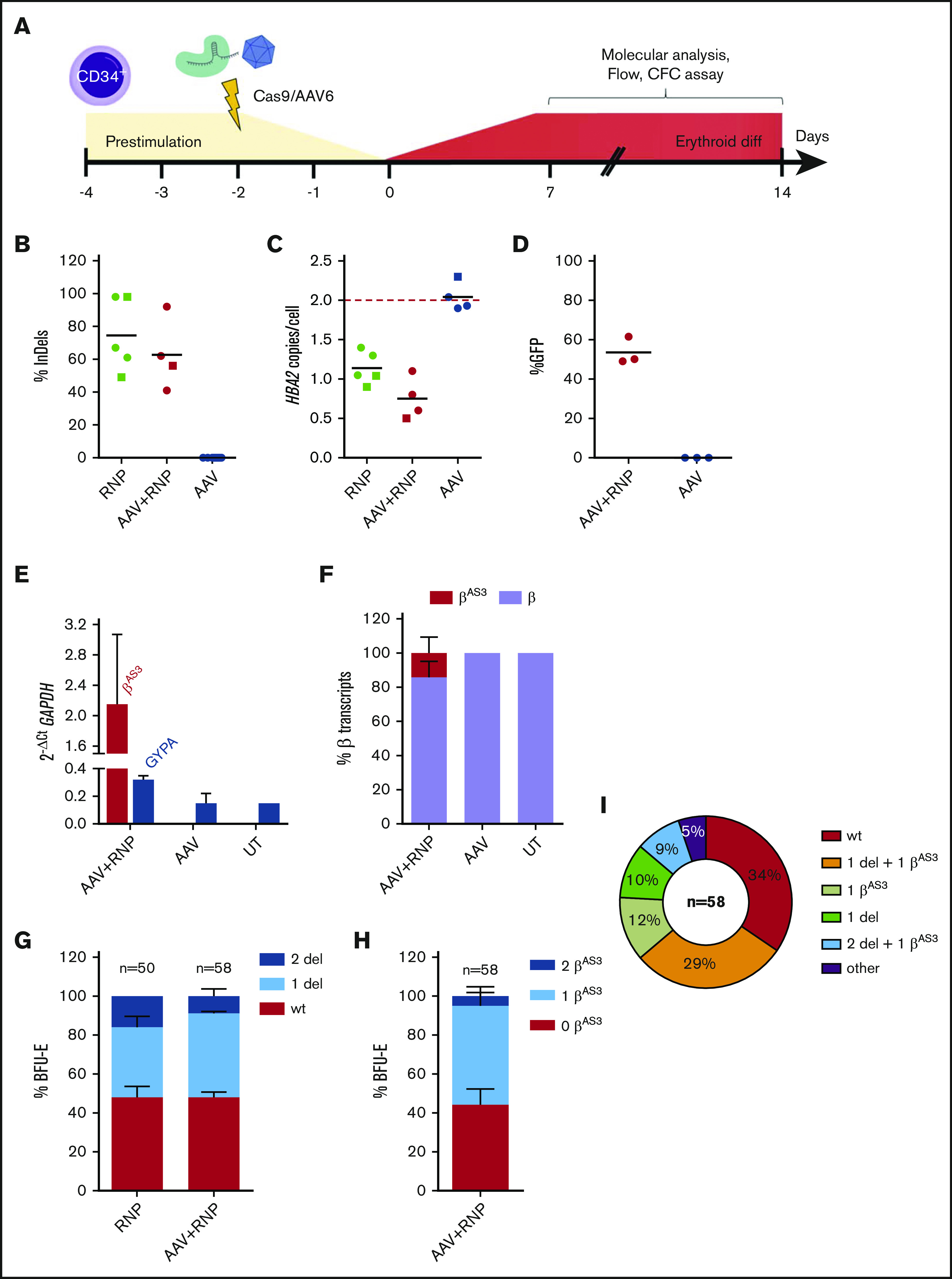
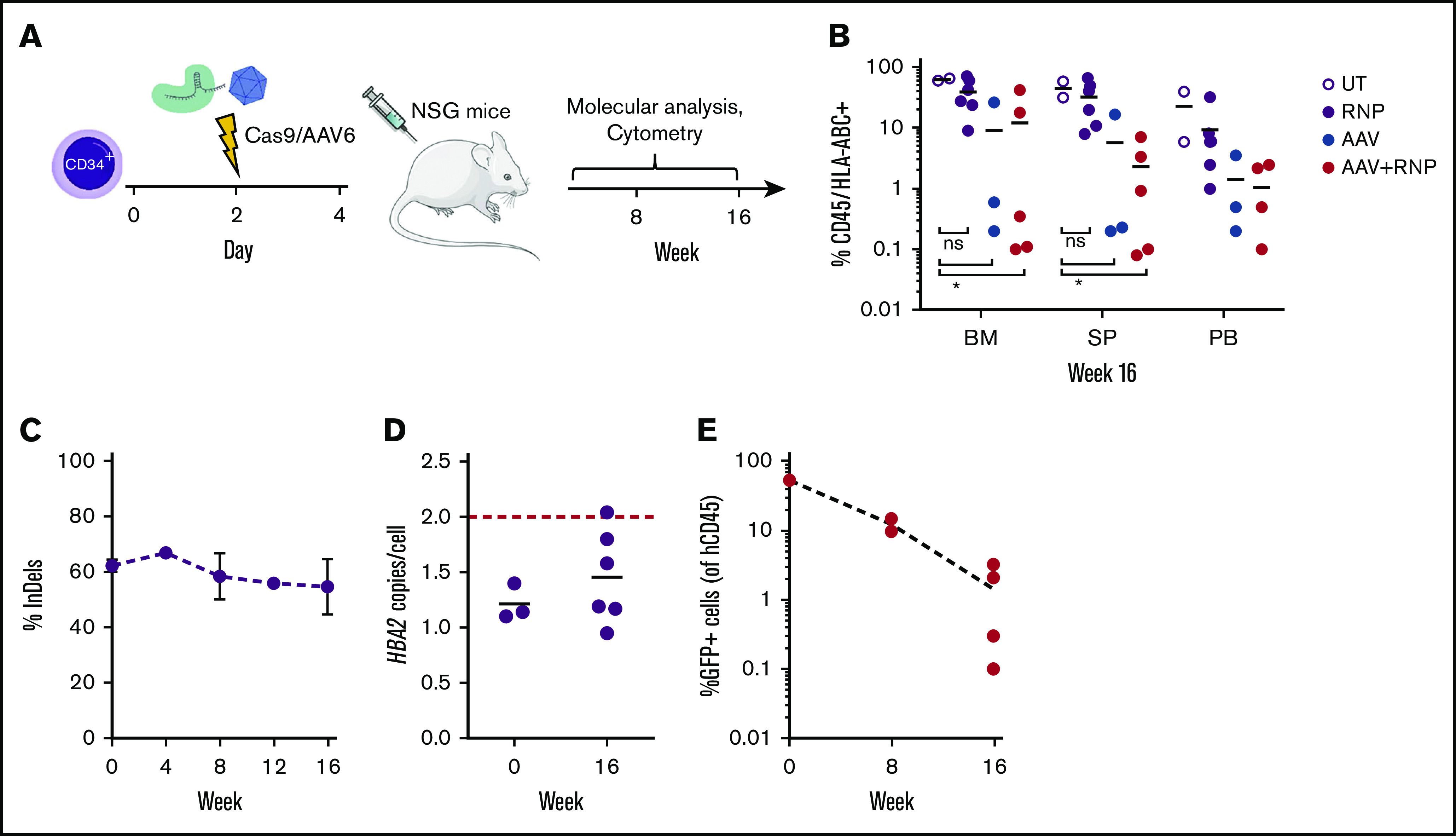
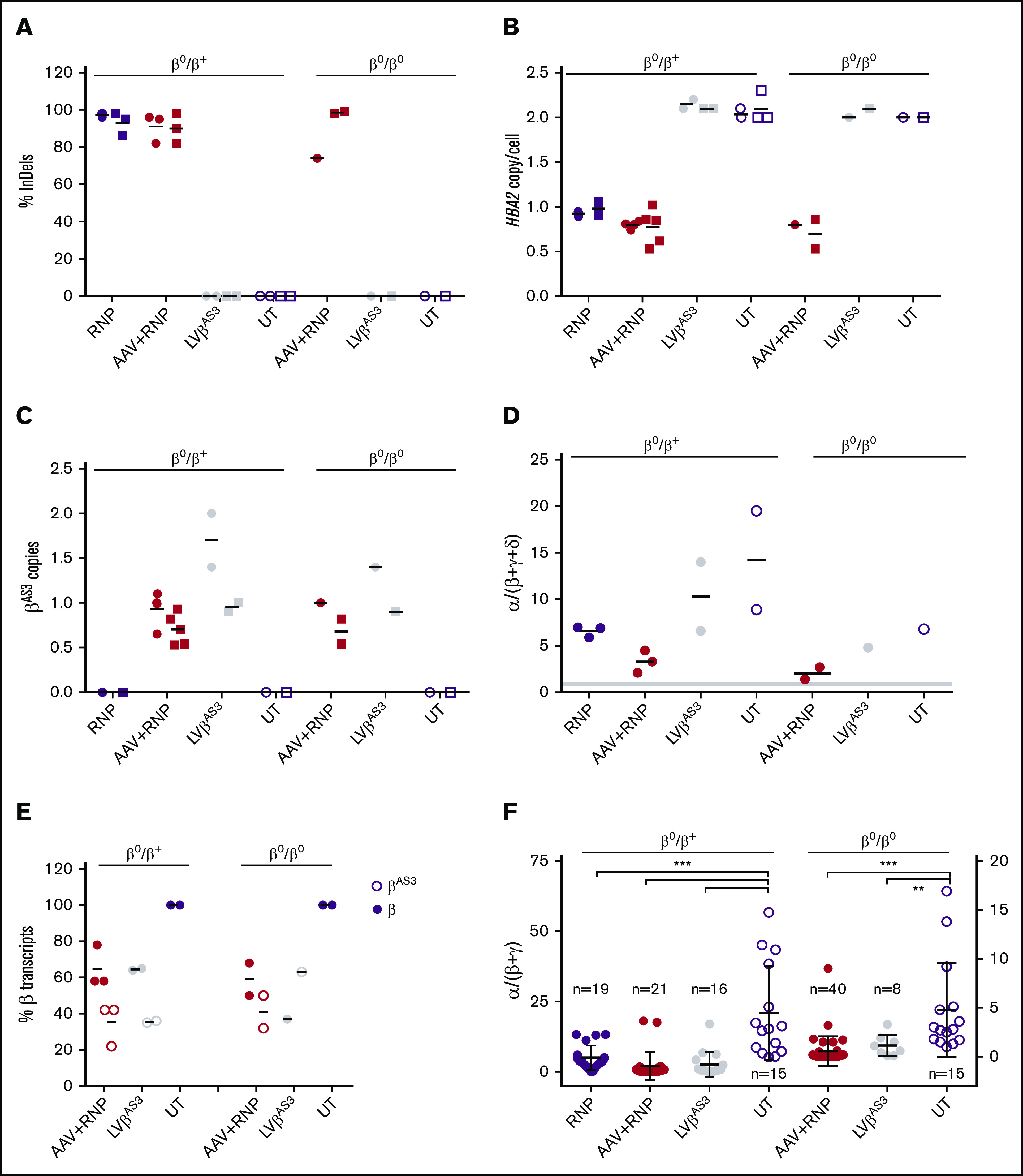
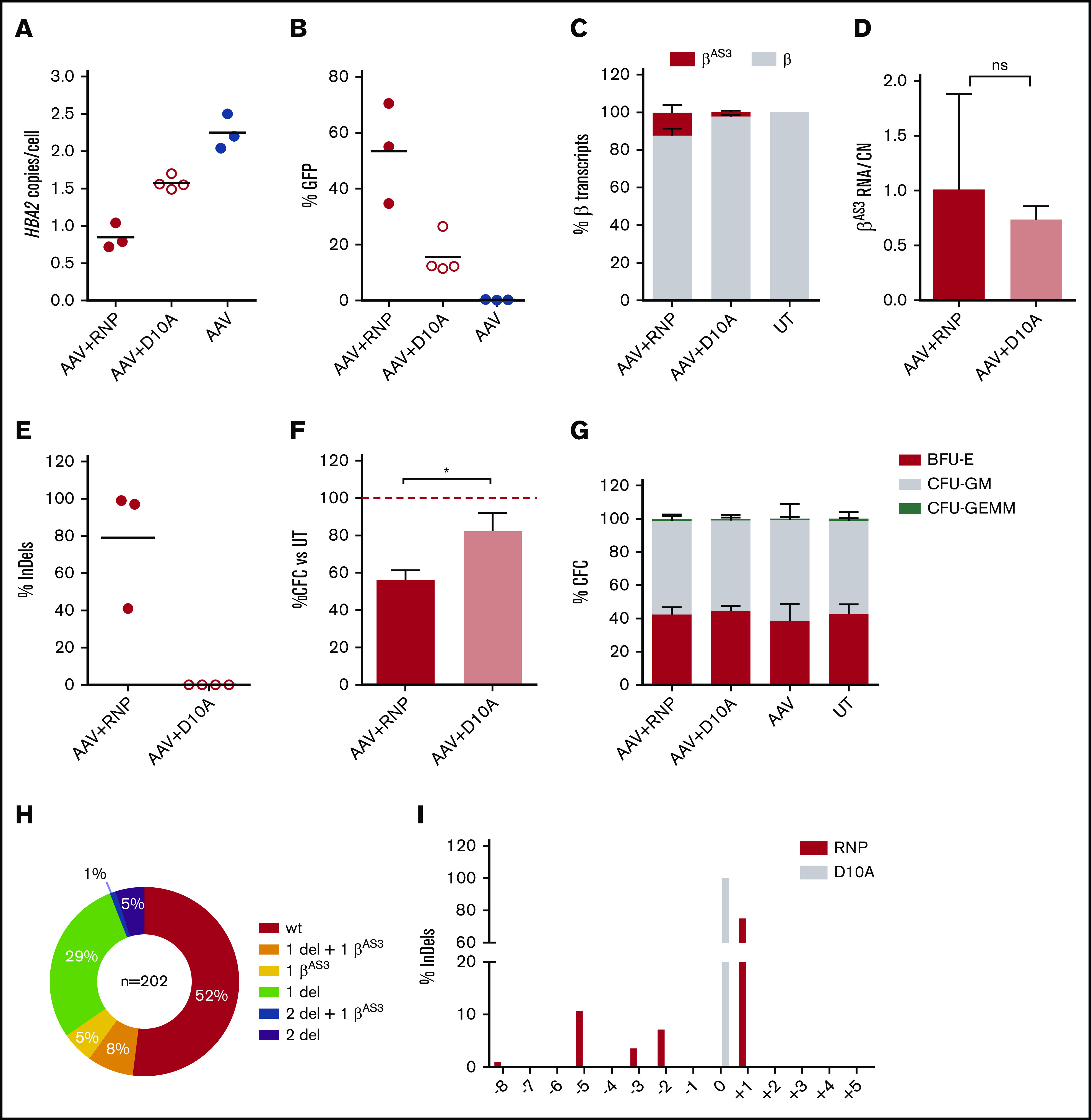
β-thalassemias (β-thal) are a group of blood disorders caused by mutations in the β-globin gene (HBB) cluster. β-globin associates with α-globin to form adult hemoglobin (HbA, α2β2), the main oxygen-carrier in erythrocytes. When β-globin chains are absent or limiting, free α-globins precipitate and damage cell membranes, causing hemolysis and ineffective erythropoiesis. Clinical data show that severity of β-thal correlates with the number of inherited α-globin genes (HBA1 and HBA2), with α-globin gene deletions having a beneficial effect for patients. Here, we describe a novel strategy to treat β-thal based on genome editing of the α-globin locus in human hematopoietic stem/progenitor cells (HSPCs). Using CRISPR/Cas9, we combined 2 therapeutic approaches: (1) α-globin downregulation, by deleting the HBA2 gene to recreate an α-thalassemia trait, and (2) β-globin expression, by targeted integration of a β-globin transgene downstream the HBA2 promoter. First, we optimized the CRISPR/Cas9 strategy and corrected the pathological phenotype in a cellular model of β-thalassemia (human erythroid progenitor cell [HUDEP-2] β0). Then, we edited healthy donor HSPCs and demonstrated that they maintained long-term repopulation capacity and multipotency in xenotransplanted mice. To assess the clinical potential of this approach, we next edited β-thal HSPCs and achieved correction of α/β globin imbalance in HSPC-derived erythroblasts. As a safer option for clinical translation, we performed editing in HSPCs using Cas9 nickase showing precise editing with no InDels. Overall, we described an innovative CRISPR/Cas9 approach to improve α/β globin imbalance in thalassemic HSPCs, paving the way for novel therapeutic strategies for β-thal.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: G.P. and M.A. are the inventors of a patent describing this HSC-based gene therapy strategy for treating β-thalassemia (correction of β-thalassemia phenotype by genetically engineering hematopoietic stem cell; EP19305484.8). The remaining authors declare no competing financial interests.
Figures
References
-
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76. - PubMed
-
- Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol. 2011;154(6):736-744. - PubMed
-
- Sharma V, Saxena R. Effect of alpha-gene numbers on phenotype of HbE/beta thalassemia patients. Ann Hematol. 2009;88(10):1035-1036. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
