

Ancestral haplotype reconstruction in endogamous populations using identity-by-descent
- PMID: 33635861
- PMCID: PMC7946327
- DOI: 10.1371/journal.pcbi.1008638
Ancestral haplotype reconstruction in endogamous populations using identity-by-descent
Abstract
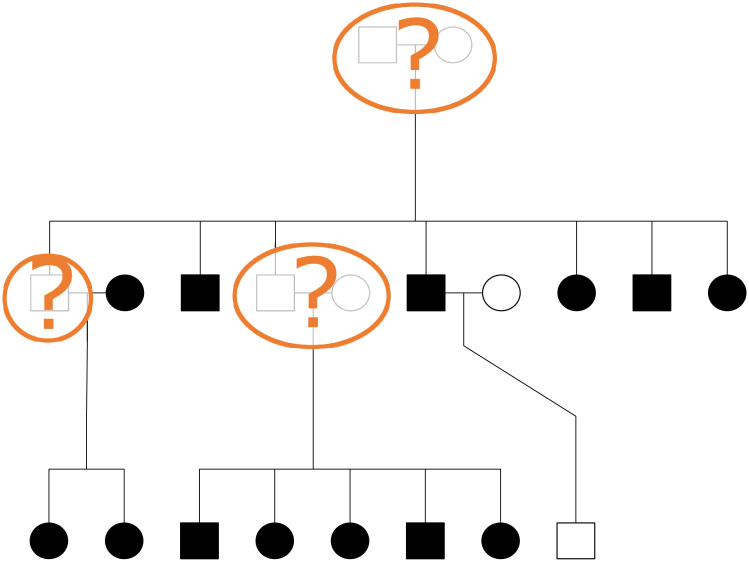
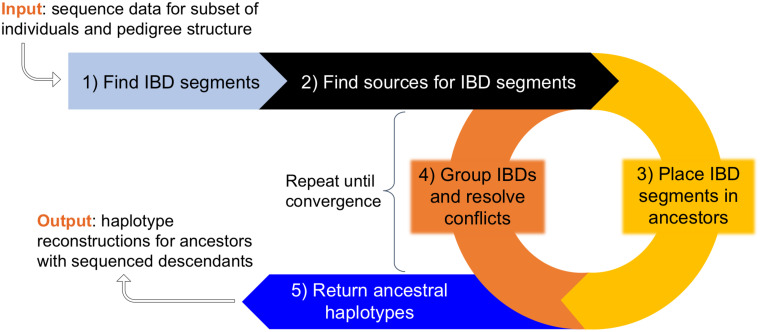
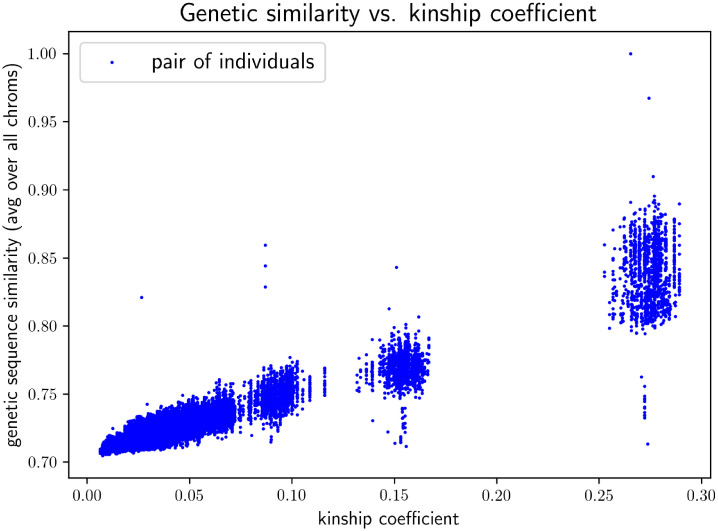
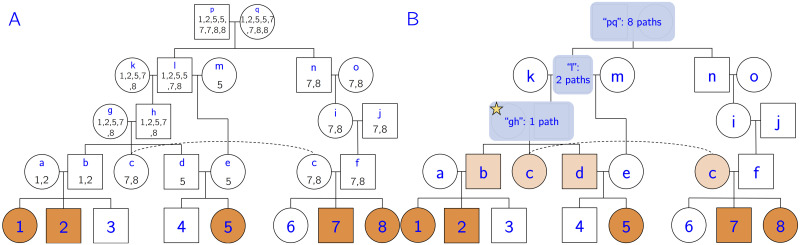
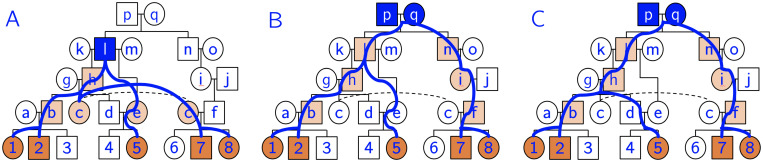
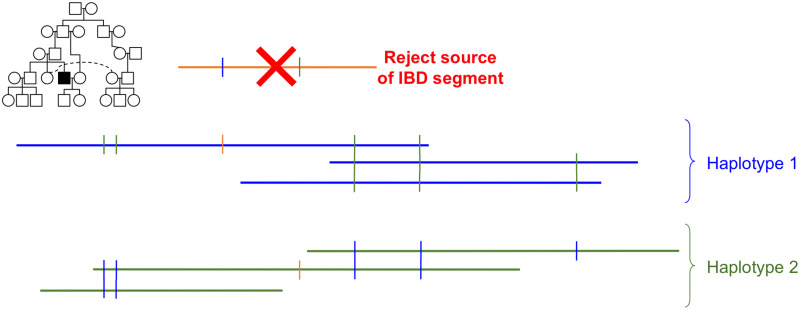
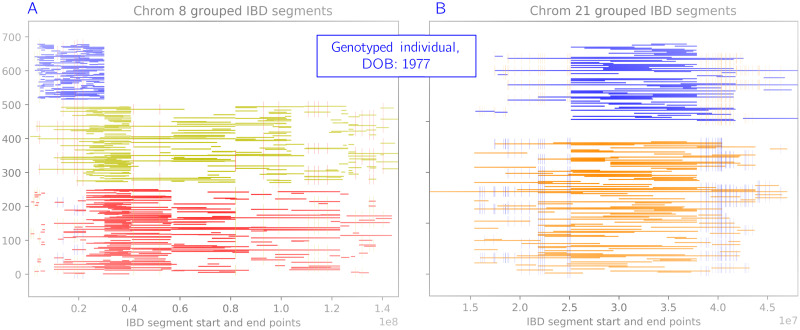
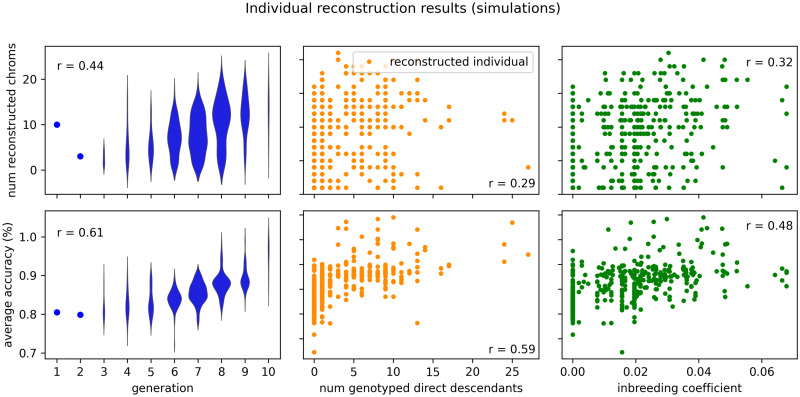
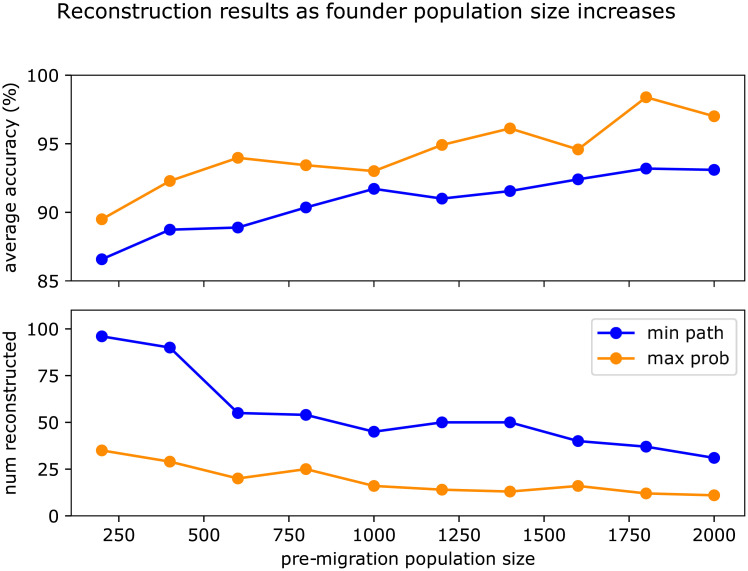
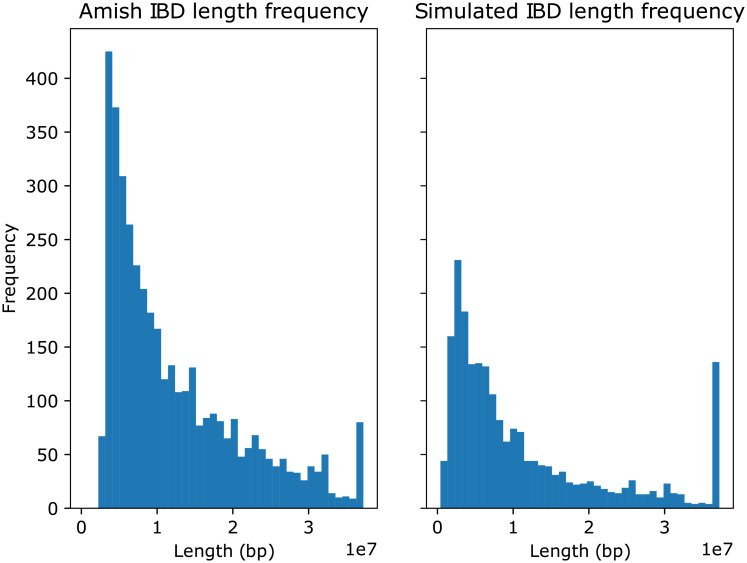
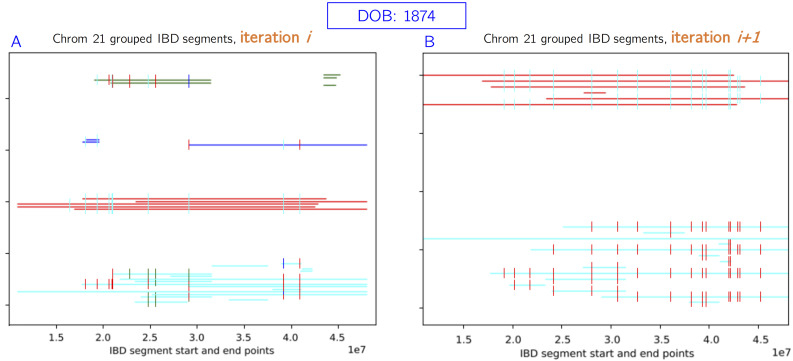
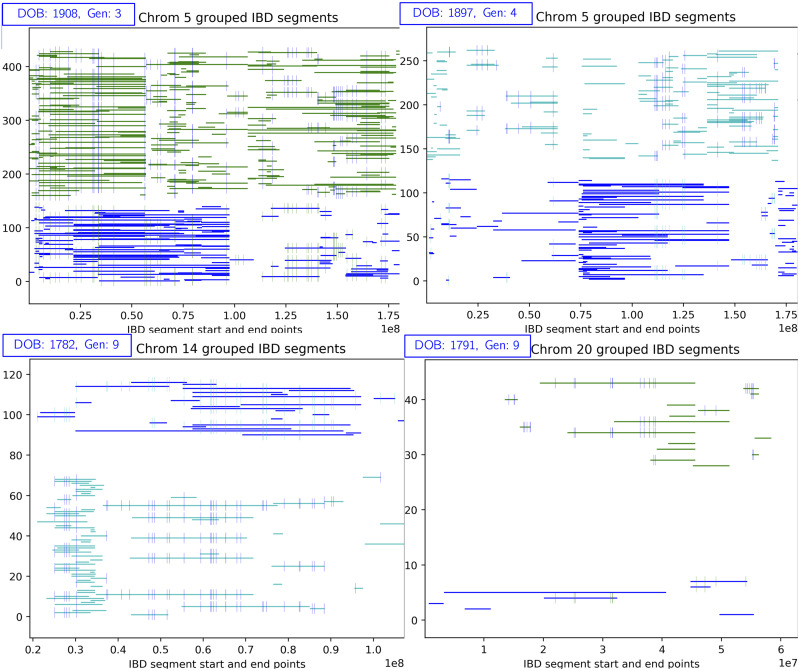
In this work we develop a novel algorithm for reconstructing the genomes of ancestral individuals, given genotype or sequence data from contemporary individuals and an extended pedigree of family relationships. A pedigree with complete genomes for every individual enables the study of allele frequency dynamics and haplotype diversity across generations, including deviations from neutrality such as transmission distortion. When studying heritable diseases, ancestral haplotypes can be used to augment genome-wide association studies and track disease inheritance patterns. The building blocks of our reconstruction algorithm are segments of Identity-By-Descent (IBD) shared between two or more genotyped individuals. The method alternates between identifying a source for each IBD segment and assembling IBD segments placed within each ancestral individual. Unlike previous approaches, our method is able to accommodate complex pedigree structures with hundreds of individuals genotyped at millions of SNPs. We apply our method to an Old Order Amish pedigree from Lancaster, Pennsylvania, whose founders came to North America from Europe during the early 18th century. The pedigree includes 1338 individuals from the past 12 generations, 394 with genotype data. The motivation for reconstruction is to understand the genetic basis of diseases segregating in the family through tracking haplotype transmission over time. Using our algorithm thread, we are able to reconstruct an average of 224 ancestral individuals per chromosome. For these ancestral individuals, on average we reconstruct 79% of their haplotypes. We also identify a region on chromosome 16 that is difficult to reconstruct-we find that this region harbors a short Amish-specific copy number variation and the gene HYDIN. thread was developed for endogamous populations, but can be applied to any extensive pedigree with the recent generations genotyped. We anticipate that this type of practical ancestral reconstruction will become more common and necessary to understand rare and complex heritable diseases in extended families.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Fast and Accurate Shared Segment Detection and Relatedness Estimation in Un-phased Genetic Data via TRUFFLE.Am J Hum Genet. 2019 Jul 3;105(1):78-88. doi: 10.1016/j.ajhg.2019.05.007. Epub 2019 Jun 6. Am J Hum Genet. 2019. PMID: 31178127 Free PMC article.
-
HAPLORE: a program for haplotype reconstruction in general pedigrees without recombination.Bioinformatics. 2005 Jan 1;21(1):90-103. doi: 10.1093/bioinformatics/bth388. Epub 2004 Jul 1. Bioinformatics. 2005. PMID: 15231536
-
Identity by descent between distant relatives: detection and applications.Annu Rev Genet. 2012;46:617-33. doi: 10.1146/annurev-genet-110711-155534. Epub 2012 Sep 17. Annu Rev Genet. 2012. PMID: 22994355 Review.
-
Whole genome identity-by-descent determination.J Bioinform Comput Biol. 2013 Apr;11(2):1350002. doi: 10.1142/S0219720013500029. Epub 2013 Jan 16. J Bioinform Comput Biol. 2013. PMID: 23600820
-
Using haplotype blocks to map human complex trait loci.Trends Genet. 2003 Mar;19(3):135-40. doi: 10.1016/S0168-9525(03)00022-2. Trends Genet. 2003. PMID: 12615007 Review.
Cited by
-
Computational Genomics and Its Applications to Anthropological Questions.Am J Biol Anthropol. 2024 Dec;186 Suppl 78(Suppl 78):e70010. doi: 10.1002/ajpa.70010. Am J Biol Anthropol. 2024. PMID: 40071816 Free PMC article. Review.
-
Long Runs of Homozygosity Are Correlated with Marriage Preferences across Global Population Samples.Hum Biol. 2021 Summer;93(3):201-216. doi: 10.1353/hub.2021.0011. Hum Biol. 2021. PMID: 37701498 Free PMC article.
-
Reconstructing parent genomes using siblings and other relatives.bioRxiv [Preprint]. 2024 May 14:2024.05.10.593578. doi: 10.1101/2024.05.10.593578. bioRxiv. 2024. PMID: 38798596 Free PMC article. Preprint.
-
Fast and Robust Identity-by-Descent Inference with the Templated Positional Burrows-Wheeler Transform.Mol Biol Evol. 2021 May 4;38(5):2131-2151. doi: 10.1093/molbev/msaa328. Mol Biol Evol. 2021. PMID: 33355662 Free PMC article.
-
Fully exploiting SNP arrays: a systematic review on the tools to extract underlying genomic structure.Brief Bioinform. 2022 Mar 10;23(2):bbac043. doi: 10.1093/bib/bbac043. Brief Bioinform. 2022. PMID: 35211719 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
