

Generating functional protein variants with variational autoencoders
- PMID: 33635868
- PMCID: PMC7946179
- DOI: 10.1371/journal.pcbi.1008736
Generating functional protein variants with variational autoencoders
Abstract
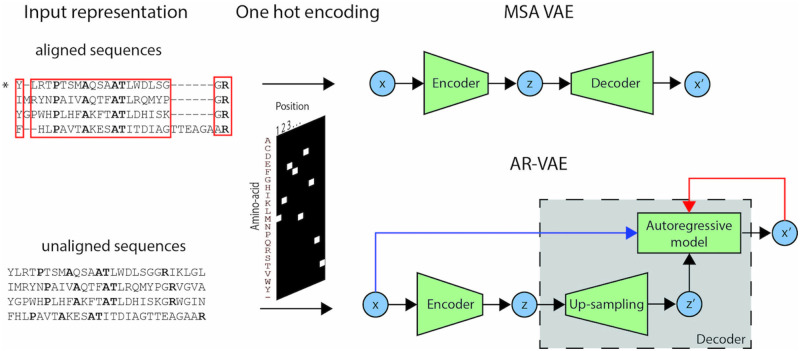
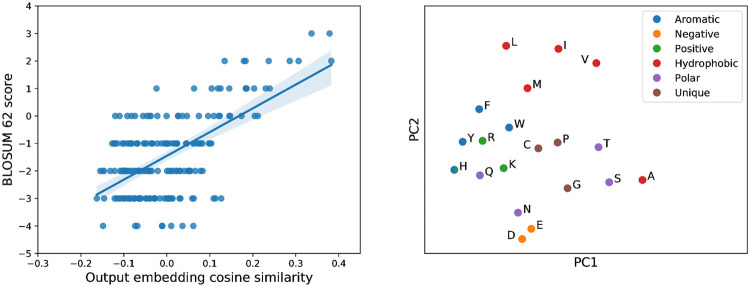
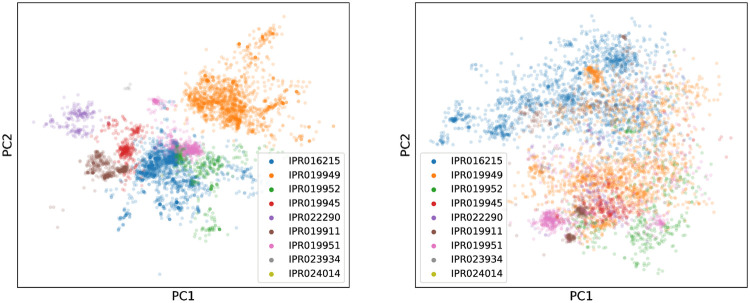
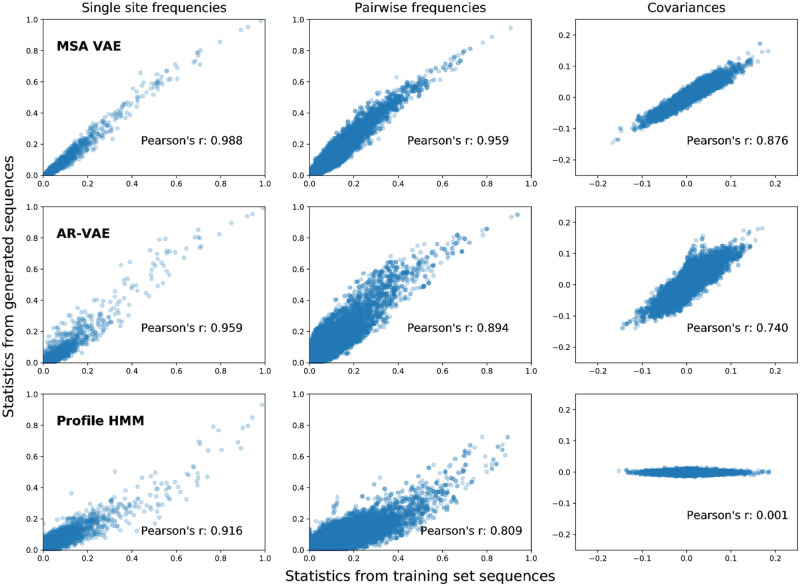
The vast expansion of protein sequence databases provides an opportunity for new protein design approaches which seek to learn the sequence-function relationship directly from natural sequence variation. Deep generative models trained on protein sequence data have been shown to learn biologically meaningful representations helpful for a variety of downstream tasks, but their potential for direct use in the design of novel proteins remains largely unexplored. Here we show that variational autoencoders trained on a dataset of almost 70000 luciferase-like oxidoreductases can be used to generate novel, functional variants of the luxA bacterial luciferase. We propose separate VAE models to work with aligned sequence input (MSA VAE) and raw sequence input (AR-VAE), and offer evidence that while both are able to reproduce patterns of amino acid usage characteristic of the family, the MSA VAE is better able to capture long-distance dependencies reflecting the influence of 3D structure. To confirm the practical utility of the models, we used them to generate variants of luxA whose luminescence activity was validated experimentally. We further showed that conditional variants of both models could be used to increase the solubility of luxA without disrupting function. Altogether 6/12 of the variants generated using the unconditional AR-VAE and 9/11 generated using the unconditional MSA VAE retained measurable luminescence, together with all 23 of the less distant variants generated by conditional versions of the models; the most distant functional variant contained 35 differences relative to the nearest training set sequence. These results demonstrate the feasibility of using deep generative models to explore the space of possible protein sequences and generate useful variants, providing a method complementary to rational design and directed evolution approaches.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
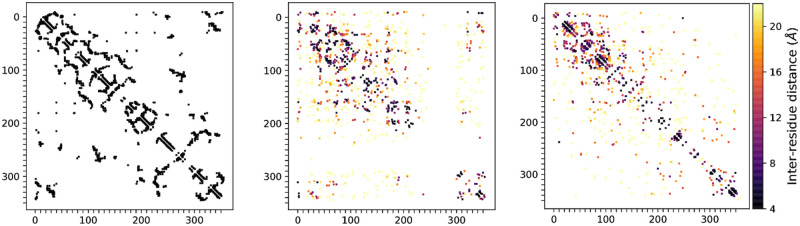
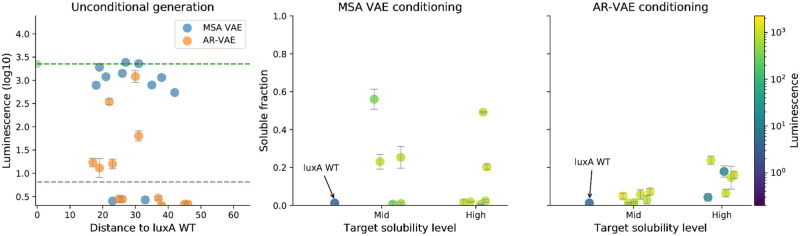
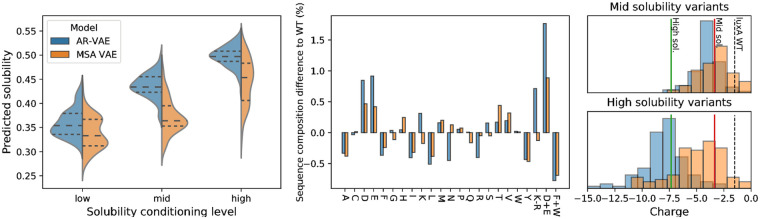
Similar articles
-
ProtWave-VAE: Integrating Autoregressive Sampling with Latent-Based Inference for Data-Driven Protein Design.ACS Synth Biol. 2023 Dec 15;12(12):3544-3561. doi: 10.1021/acssynbio.3c00261. Epub 2023 Nov 21. ACS Synth Biol. 2023. PMID: 37988083 Free PMC article.
-
A hybrid variational autoencoder and WGAN with gradient penalty for tertiary protein structure generation.Sci Rep. 2025 Apr 23;15(1):14191. doi: 10.1038/s41598-025-94747-y. Sci Rep. 2025. PMID: 40268976 Free PMC article.
-
Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders.Pac Symp Biocomput. 2018;23:80-91. Pac Symp Biocomput. 2018. PMID: 29218871 Free PMC article.
-
An Overview of Variational Autoencoders for Source Separation, Finance, and Bio-Signal Applications.Entropy (Basel). 2021 Dec 28;24(1):55. doi: 10.3390/e24010055. Entropy (Basel). 2021. PMID: 35052081 Free PMC article. Review.
-
Generative models for protein sequence modeling: recent advances and future directions.Brief Bioinform. 2023 Sep 22;24(6):bbad358. doi: 10.1093/bib/bbad358. Brief Bioinform. 2023. PMID: 37864295 Free PMC article. Review.
Cited by
-
Funneling modulatory peptide design with generative models: Discovery and characterization of disruptors of calcineurin protein-protein interactions.PLoS Comput Biol. 2023 Feb 2;19(2):e1010874. doi: 10.1371/journal.pcbi.1010874. eCollection 2023 Feb. PLoS Comput Biol. 2023. PMID: 36730443 Free PMC article.
-
Therapeutic enzyme engineering using a generative neural network.Sci Rep. 2022 Jan 27;12(1):1536. doi: 10.1038/s41598-022-05195-x. Sci Rep. 2022. PMID: 35087131 Free PMC article.
-
Engineering Dehalogenase Enzymes Using Variational Autoencoder-Generated Latent Spaces and Microfluidics.JACS Au. 2025 Feb 13;5(2):838-850. doi: 10.1021/jacsau.4c01101. eCollection 2025 Feb 24. JACS Au. 2025. PMID: 40017771 Free PMC article.
-
Bayesian estimation of muscle mechanisms and therapeutic targets using variational autoencoders.Biophys J. 2025 Jan 7;124(1):179-191. doi: 10.1016/j.bpj.2024.11.3310. Epub 2024 Nov 26. Biophys J. 2025. PMID: 39604261 Free PMC article.
-
AMPGAN v2: Machine Learning-Guided Design of Antimicrobial Peptides.J Chem Inf Model. 2021 May 24;61(5):2198-2207. doi: 10.1021/acs.jcim.0c01441. Epub 2021 Mar 31. J Chem Inf Model. 2021. PMID: 33787250 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
