

Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis
- PMID: 33636128
- PMCID: PMC8109274
- DOI: 10.1016/j.cell.2021.01.049
Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis
Abstract
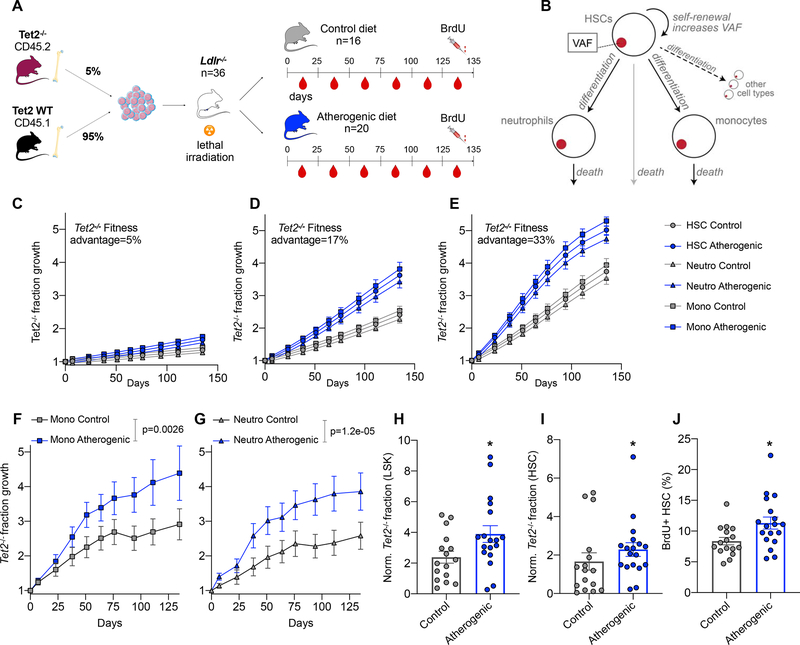
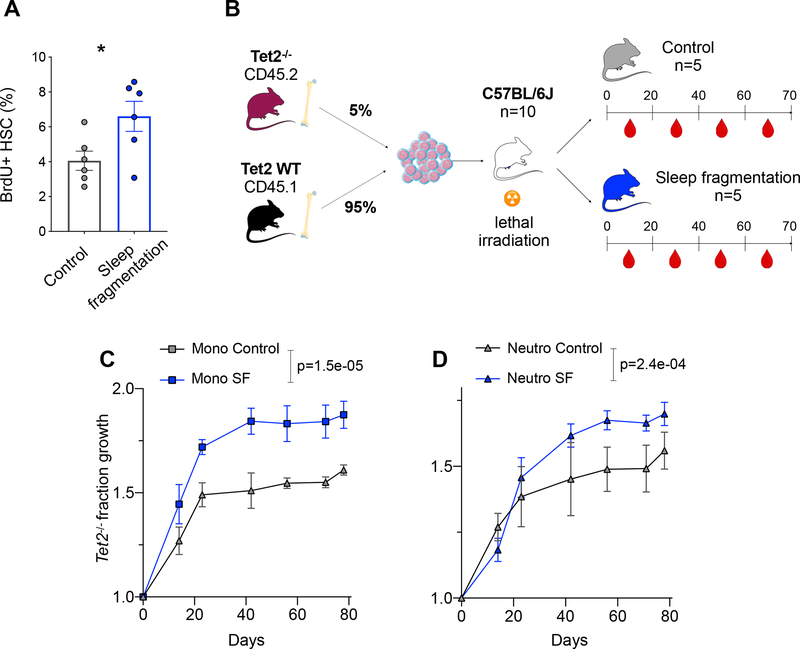
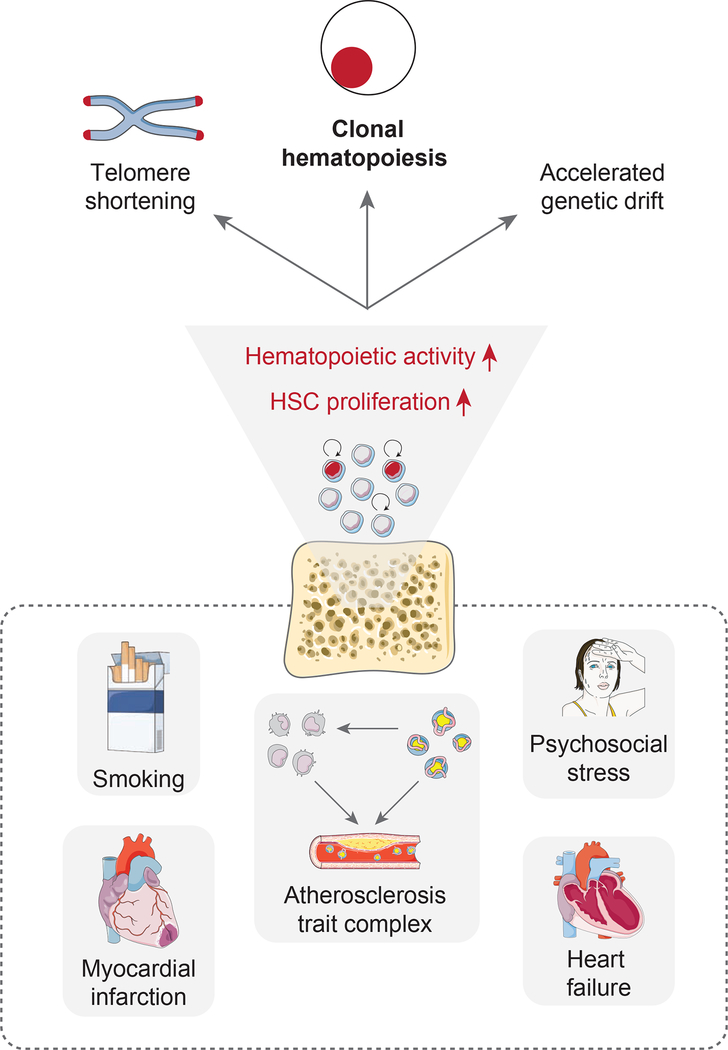
Clonal hematopoiesis, a condition in which individual hematopoietic stem cell clones generate a disproportionate fraction of blood leukocytes, correlates with higher risk for cardiovascular disease. The mechanisms behind this association are incompletely understood. Here, we show that hematopoietic stem cell division rates are increased in mice and humans with atherosclerosis. Mathematical analysis demonstrates that increased stem cell proliferation expedites somatic evolution and expansion of clones with driver mutations. The experimentally determined division rate elevation in atherosclerosis patients is sufficient to produce a 3.5-fold increased risk of clonal hematopoiesis by age 70. We confirm the accuracy of our theoretical framework in mouse models of atherosclerosis and sleep fragmentation by showing that expansion of competitively transplanted Tet2-/- cells is accelerated under conditions of chronically elevated hematopoietic activity. Hence, increased hematopoietic stem cell proliferation is an important factor contributing to the association between cardiovascular disease and clonal hematopoiesis.
Keywords: atherosclerosis; clonal hematopoiesis; hematopoietic stem cell; somatic evolution.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
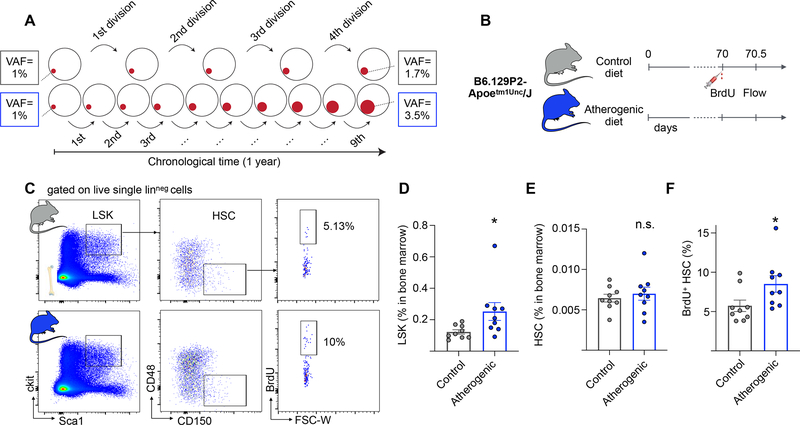
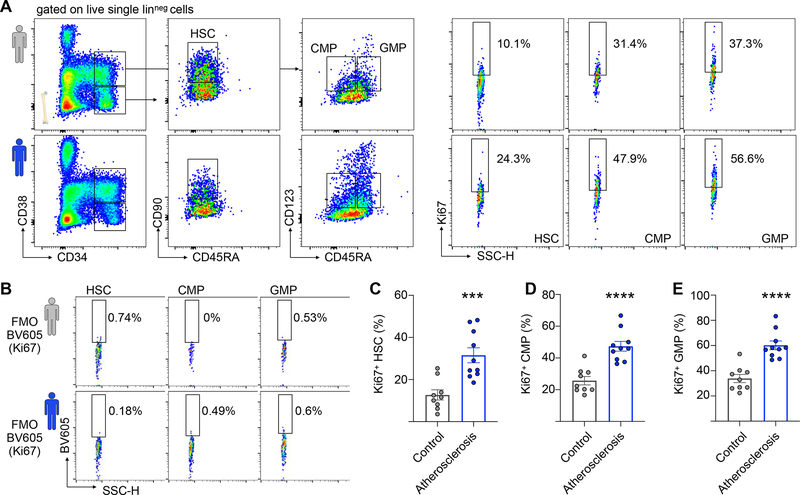
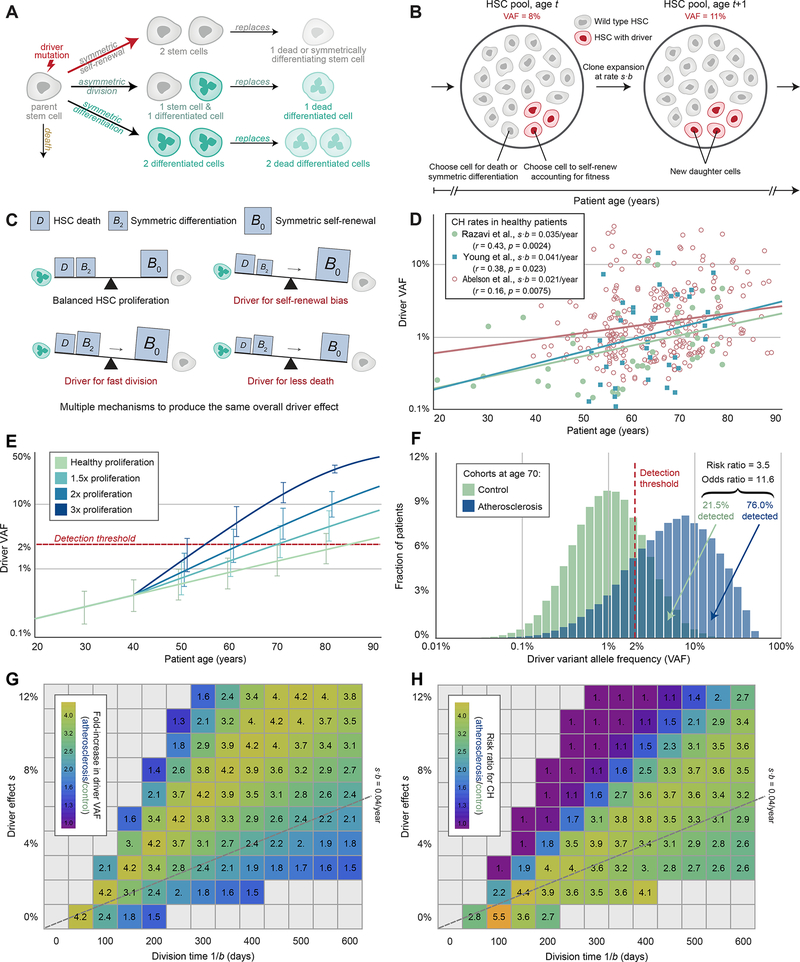
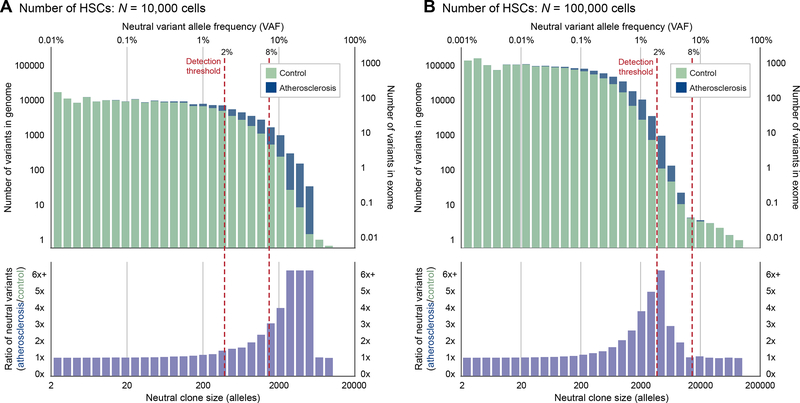
Comment in
-
A vicious cycle in atherosclerosis.Cell. 2021 Mar 4;184(5):1139-1141. doi: 10.1016/j.cell.2021.02.005. Cell. 2021. PMID: 33667367
-
Clonal haematopoiesis and atherosclerosis: a chicken or egg question?Nat Rev Cardiol. 2021 Jul;18(7):463-464. doi: 10.1038/s41569-021-00554-z. Nat Rev Cardiol. 2021. PMID: 33859398 No abstract available.
References
-
- Abkowitz JL, Golinelli D, Harrison DE, and Guttorp P (2000). In vivo kinetics of murine hemopoietic stem cells. Blood 96, 3399–3405. - PubMed
-
- Abkowitz JL, Catlin SN, McCallie MT, and Guttorp P (2002). Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood 100, 2665–2667. - PubMed
-
- Altrock PM, Liu LL, and Michor F (2015). The mathematics of cancer: integrating quantitative models. Nat. Rev. Cancer 15, 730–745. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
