

On the use of whole-genome sequence data for across-breed genomic prediction and fine-scale mapping of QTL
- PMID: 33637049
- PMCID: PMC7908738
- DOI: 10.1186/s12711-021-00607-4
On the use of whole-genome sequence data for across-breed genomic prediction and fine-scale mapping of QTL
Abstract
Background: Whole-genome sequence (WGS) data are increasingly available on large numbers of individuals in animal and plant breeding and in human genetics through second-generation resequencing technologies, 1000 genomes projects, and large-scale genotype imputation from lower marker densities. Here, we present a computationally fast implementation of a variable selection genomic prediction method, that could handle WGS data on more than 35,000 individuals, test its accuracy for across-breed predictions and assess its quantitative trait locus (QTL) mapping precision.
Methods: The Monte Carlo Markov chain (MCMC) variable selection model (Bayes GC) fits simultaneously a genomic best linear unbiased prediction (GBLUP) term, i.e. a polygenic effect whose correlations are described by a genomic relationship matrix (G), and a Bayes C term, i.e. a set of single nucleotide polymorphisms (SNPs) with large effects selected by the model. Computational speed is improved by a Metropolis-Hastings sampling that directs computations to the SNPs, which are, a priori, most likely to be included into the model. Speed is also improved by running many relatively short MCMC chains. Memory requirements are reduced by storing the genotype matrix in binary form. The model was tested on a WGS dataset containing Holstein, Jersey and Australian Red cattle. The data contained 4,809,520 genotypes on 35,549 individuals together with their milk, fat and protein yields, and fat and protein percentage traits.
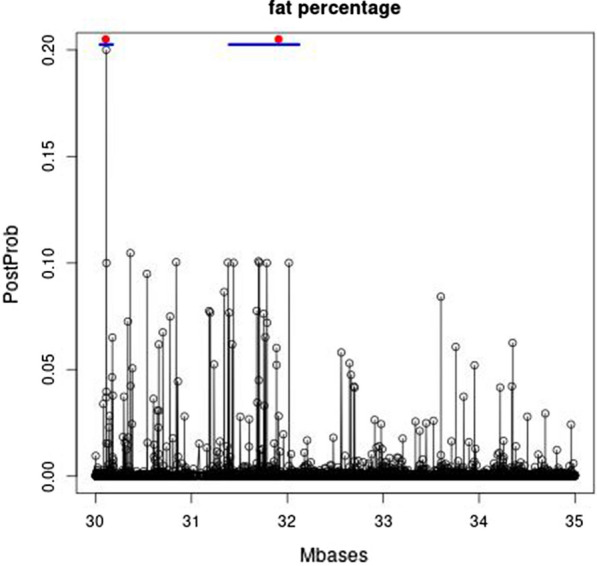
Results: The prediction accuracies of the Jersey individuals improved by 1.5% when using across-breed GBLUP compared to within-breed predictions. Using WGS instead of 600 k SNP-chip data yielded on average a 3% accuracy improvement for Australian Red cows. QTL were fine-mapped by locating the SNP with the highest posterior probability of being included in the model. Various QTL known from the literature were rediscovered, and a new SNP affecting milk production was discovered on chromosome 20 at 34.501126 Mb. Due to the high mapping precision, it was clear that many of the discovered QTL were the same across the five dairy traits.
Conclusions: Across-breed Bayes GC genomic prediction improved prediction accuracies compared to GBLUP. The combination of across-breed WGS data and Bayesian genomic prediction proved remarkably effective for the fine-mapping of QTL.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Similar articles
-
Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions.Genet Sel Evol. 2015 Apr 17;47(1):29. doi: 10.1186/s12711-014-0074-4. Genet Sel Evol. 2015. PMID: 25887988 Free PMC article.
-
Application of a Bayesian non-linear model hybrid scheme to sequence data for genomic prediction and QTL mapping.BMC Genomics. 2017 Aug 15;18(1):618. doi: 10.1186/s12864-017-4030-x. BMC Genomics. 2017. PMID: 28810831 Free PMC article.
-
Impact of QTL properties on the accuracy of multi-breed genomic prediction.Genet Sel Evol. 2015 May 8;47(1):42. doi: 10.1186/s12711-015-0124-6. Genet Sel Evol. 2015. PMID: 25951906 Free PMC article.
-
Genomic prediction in animals and plants: simulation of data, validation, reporting, and benchmarking.Genetics. 2013 Feb;193(2):347-65. doi: 10.1534/genetics.112.147983. Epub 2012 Dec 5. Genetics. 2013. PMID: 23222650 Free PMC article. Review.
-
Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans: Genomic Prediction.Genetics. 2019 Apr;211(4):1131-1141. doi: 10.1534/genetics.119.301859. Genetics. 2019. PMID: 30967442 Free PMC article. Review.
Cited by
-
Multi-breed genomic evaluation for tropical beef cattle when no pedigree information is available.Genet Sel Evol. 2023 Oct 16;55(1):71. doi: 10.1186/s12711-023-00847-6. Genet Sel Evol. 2023. PMID: 37845626 Free PMC article.
-
Hybrid de novo and haplotype-resolved genome assembly of Vechur cattle - elucidating genetic variation.Front Genet. 2024 Mar 6;15:1338224. doi: 10.3389/fgene.2024.1338224. eCollection 2024. Front Genet. 2024. PMID: 38510276 Free PMC article.
-
Multi-line ssGBLUP evaluation using preselected markers from whole-genome sequence data in pigs.Front Genet. 2023 May 12;14:1163626. doi: 10.3389/fgene.2023.1163626. eCollection 2023. Front Genet. 2023. PMID: 37252662 Free PMC article.
-
Potential negative effects of genomic selection.J Anim Sci. 2024 Jan 3;102:skae155. doi: 10.1093/jas/skae155. J Anim Sci. 2024. PMID: 38847068 Free PMC article.
-
Genomic evaluation with multibreed and crossbred data.JDS Commun. 2022 Jan 10;3(2):156-159. doi: 10.3168/jdsc.2021-0177. eCollection 2022 Mar. JDS Commun. 2022. PMID: 36339739 Free PMC article. Review.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
