

Targeted de-repression of neuronal Nrf2 inhibits α-synuclein accumulation
- PMID: 33637689
- PMCID: PMC7910424
- DOI: 10.1038/s41419-021-03507-z
Targeted de-repression of neuronal Nrf2 inhibits α-synuclein accumulation
Abstract
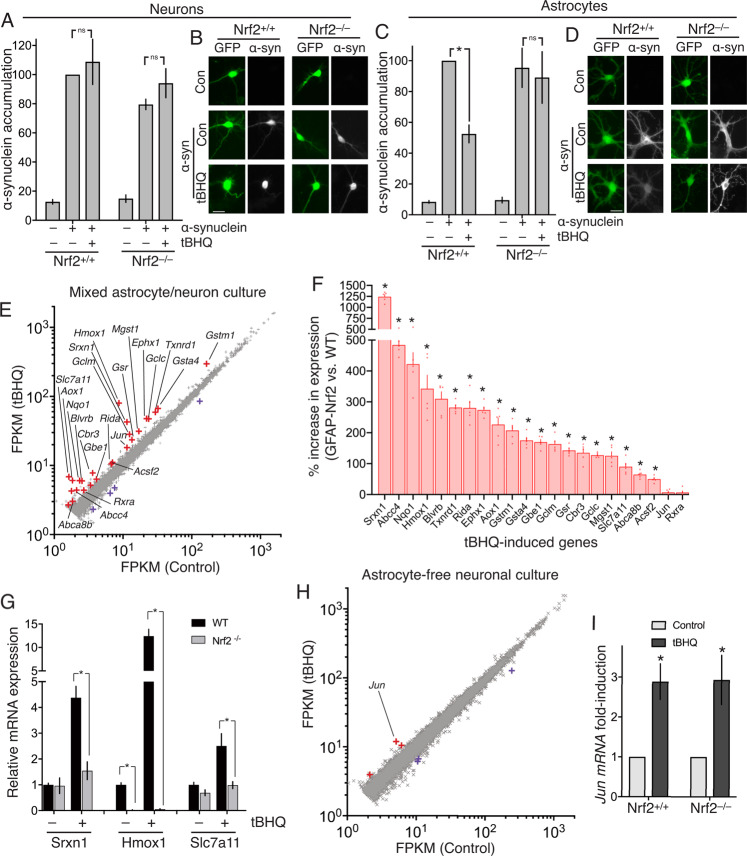
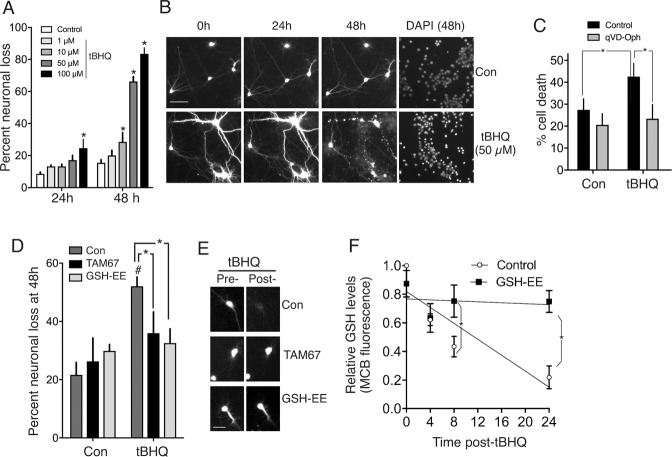
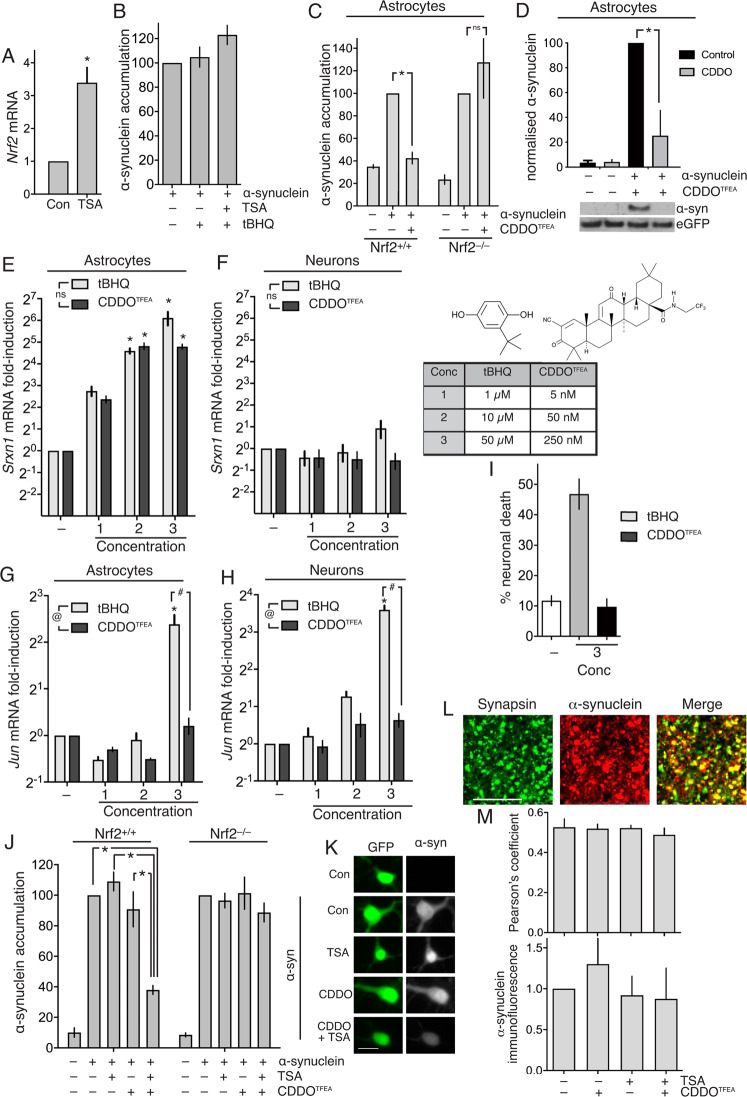
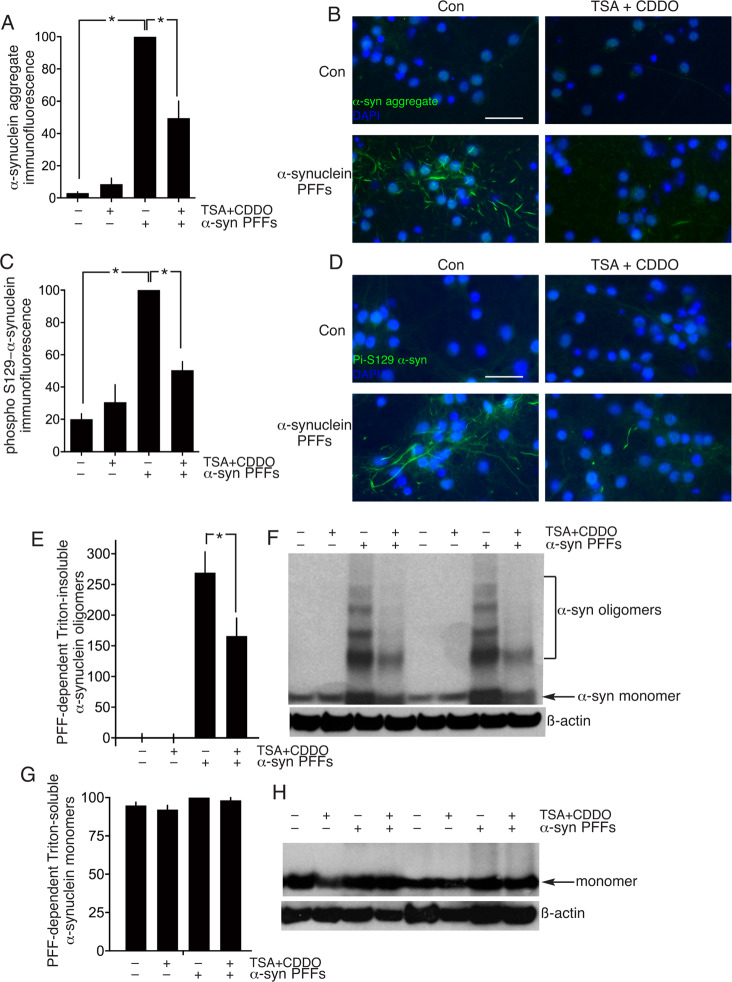
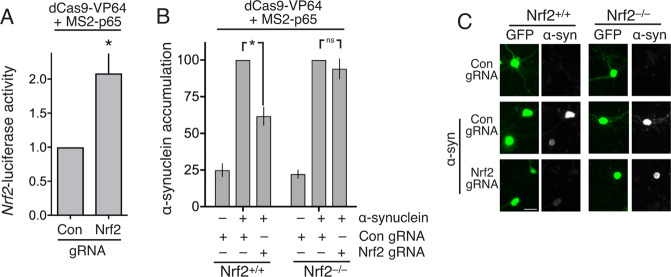
Many neurodegenerative diseases are associated with neuronal misfolded protein accumulation, indicating a need for proteostasis-promoting strategies. Here we show that de-repressing the transcription factor Nrf2, epigenetically shut-off in early neuronal development, can prevent protein aggregate accumulation. Using a paradigm of α-synuclein accumulation and clearance, we find that the classical electrophilic Nrf2 activator tBHQ promotes endogenous Nrf2-dependent α-synuclein clearance in astrocytes, but not cortical neurons, which mount no Nrf2-dependent transcriptional response. Moreover, due to neuronal Nrf2 shut-off and consequent weak antioxidant defences, electrophilic tBHQ actually induces oxidative neurotoxicity, via Nrf2-independent Jun induction. However, we find that epigenetic de-repression of neuronal Nrf2 enables them to respond to Nrf2 activators to drive α-synuclein clearance. Moreover, activation of neuronal Nrf2 expression using gRNA-targeted dCas9-based transcriptional activation complexes is sufficient to trigger Nrf2-dependent α-synuclein clearance. Thus, targeting reversal of the developmental shut-off of Nrf2 in forebrain neurons may alter neurodegenerative disease trajectory by boosting proteostasis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Tebay LE, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015;88(Pt B):108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
