

Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome
- PMID: 33637765
- PMCID: PMC7910481
- DOI: 10.1038/s41467-021-21588-4
Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome
Abstract
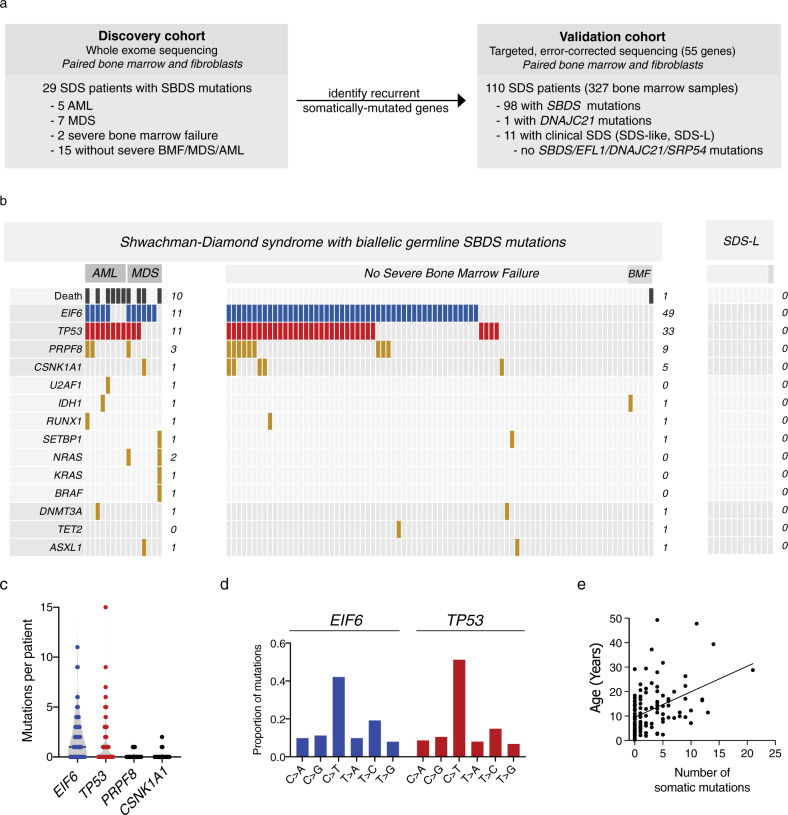
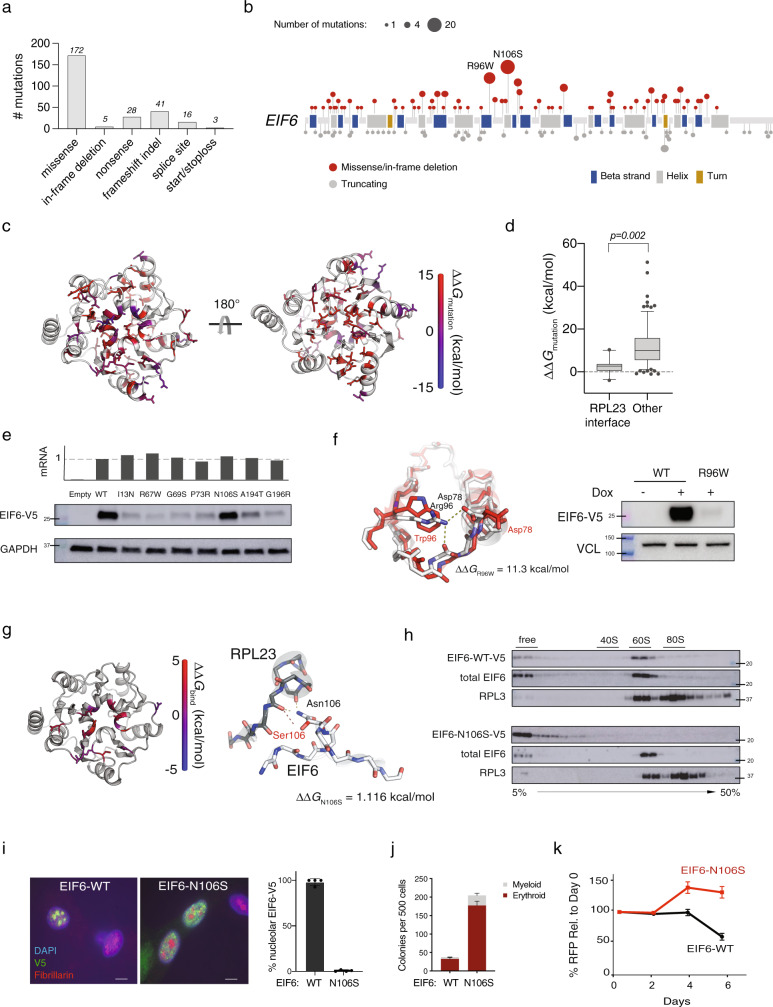
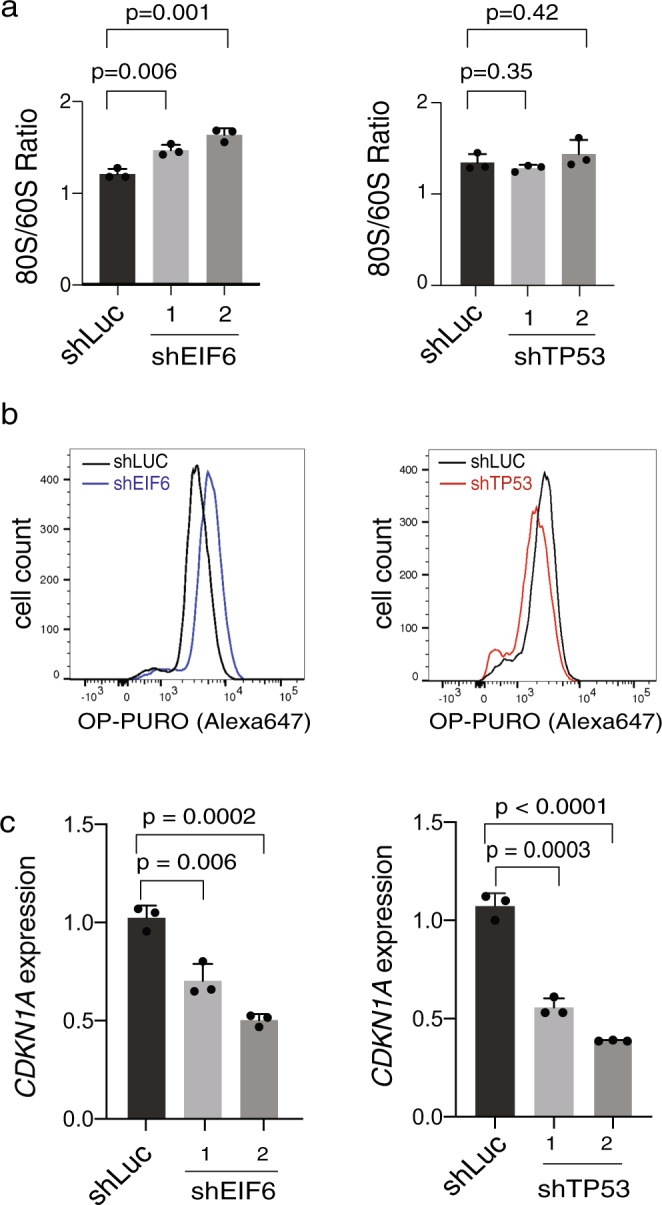
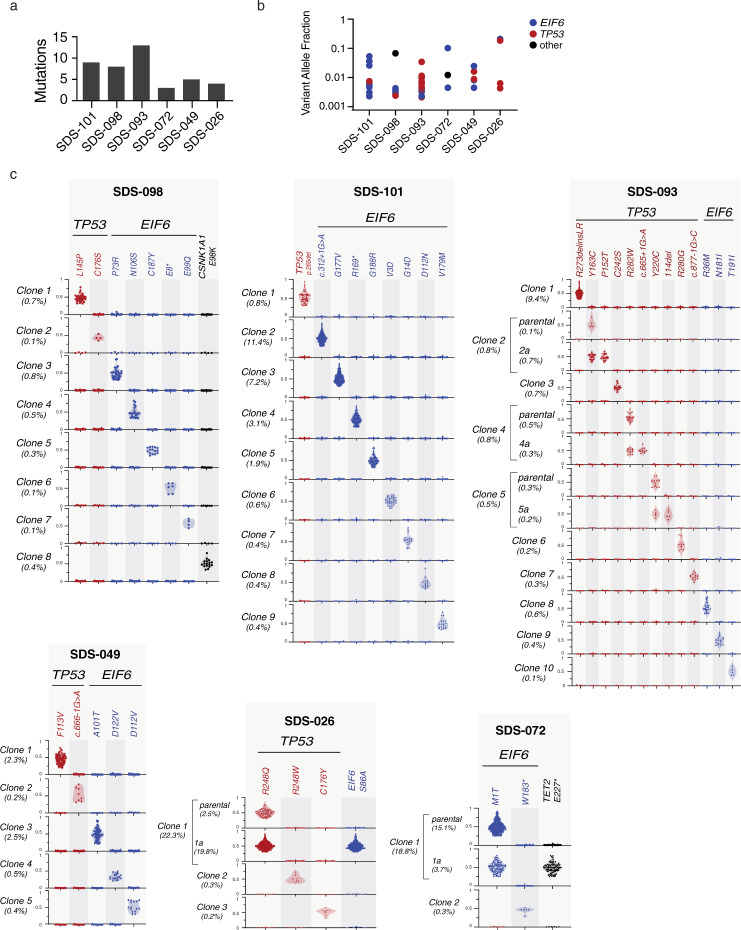
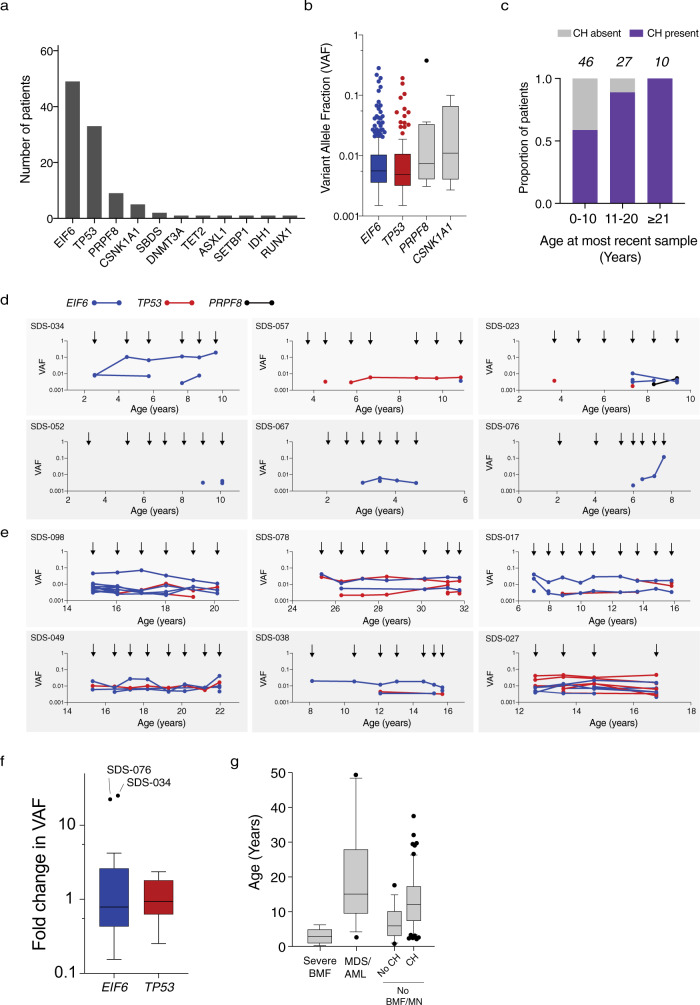
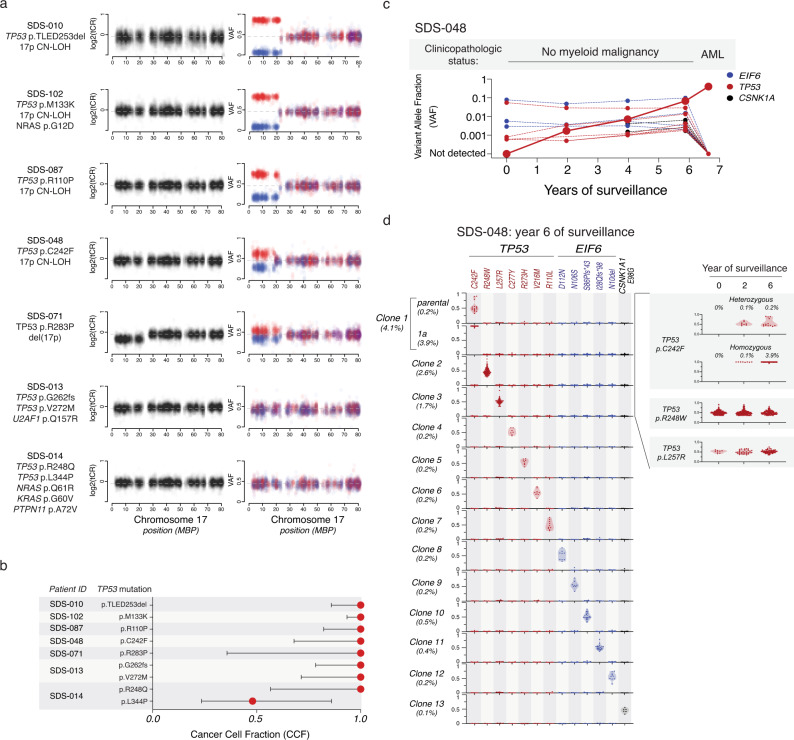
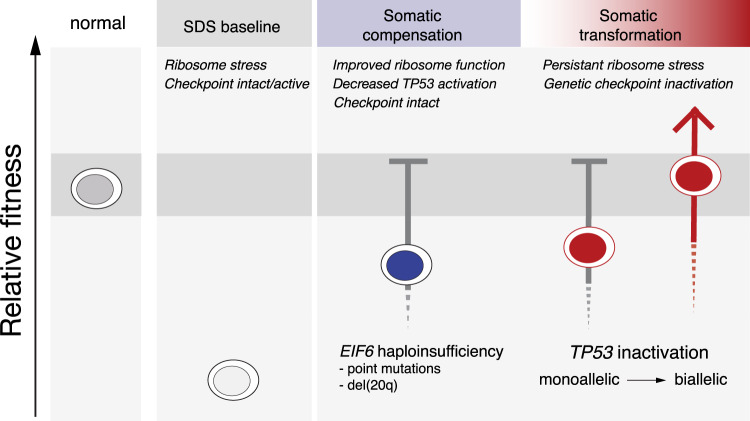
To understand the mechanisms that mediate germline genetic leukemia predisposition, we studied the inherited ribosomopathy Shwachman-Diamond syndrome (SDS), a bone marrow failure disorder with high risk of myeloid malignancies at an early age. To define the mechanistic basis of clonal hematopoiesis in SDS, we investigate somatic mutations acquired by patients with SDS followed longitudinally. Here we report that multiple independent somatic hematopoietic clones arise early in life, most commonly harboring heterozygous mutations in EIF6 or TP53. We show that germline SBDS deficiency establishes a fitness constraint that drives selection of somatic clones via two distinct mechanisms with different clinical consequences. EIF6 inactivation mediates a compensatory pathway with limited leukemic potential by ameliorating the underlying SDS ribosome defect and enhancing clone fitness. TP53 mutations define a maladaptive pathway with enhanced leukemic potential by inactivating tumor suppressor checkpoints without correcting the ribosome defect. Subsequent development of leukemia was associated with acquisition of biallelic TP53 alterations. These results mechanistically link leukemia predisposition to germline genetic constraints on cellular fitness, and provide a rational framework for clinical surveillance strategies.
Conflict of interest statement
J.R.E. is a part of the speakers’ Bureau: Jazz Pharmaceuticals; Astellas, Consultant: Jazz Pharmaceuticals. N.H. is a consultant for Novartis and Incyte. S.M.D. is a consultant for Alexion and Novartis. R.C.L. has received research funding from Jazz Pharmaceuticals, and consulting fees from Takeda Pharmaceuticals and bluebird bio. Other authors have no competing interests to declare.
Figures
References
-
- Arber DA, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. Blood. 2016;128:462–463. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
