

Deep learning techniques have significantly impacted protein structure prediction and protein design
- PMID: 33639355
- PMCID: PMC8222070
- DOI: 10.1016/j.sbi.2021.01.007
Deep learning techniques have significantly impacted protein structure prediction and protein design
Abstract
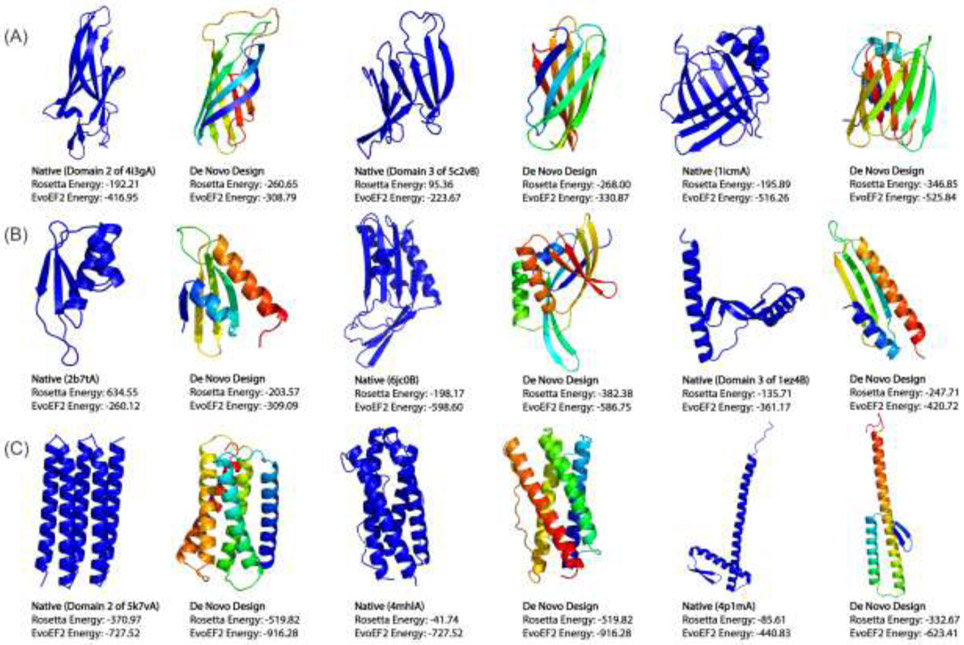
Protein structure prediction and design can be regarded as two inverse processes governed by the same folding principle. Although progress remained stagnant over the past two decades, the recent application of deep neural networks to spatial constraint prediction and end-to-end model training has significantly improved the accuracy of protein structure prediction, largely solving the problem at the fold level for single-domain proteins. The field of protein design has also witnessed dramatic improvement, where noticeable examples have shown that information stored in neural-network models can be used to advance functional protein design. Thus, incorporation of deep learning techniques into different steps of protein folding and design approaches represents an exciting future direction and should continue to have a transformative impact on both fields.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interest
The authors declare no conflict of interest.
Figures
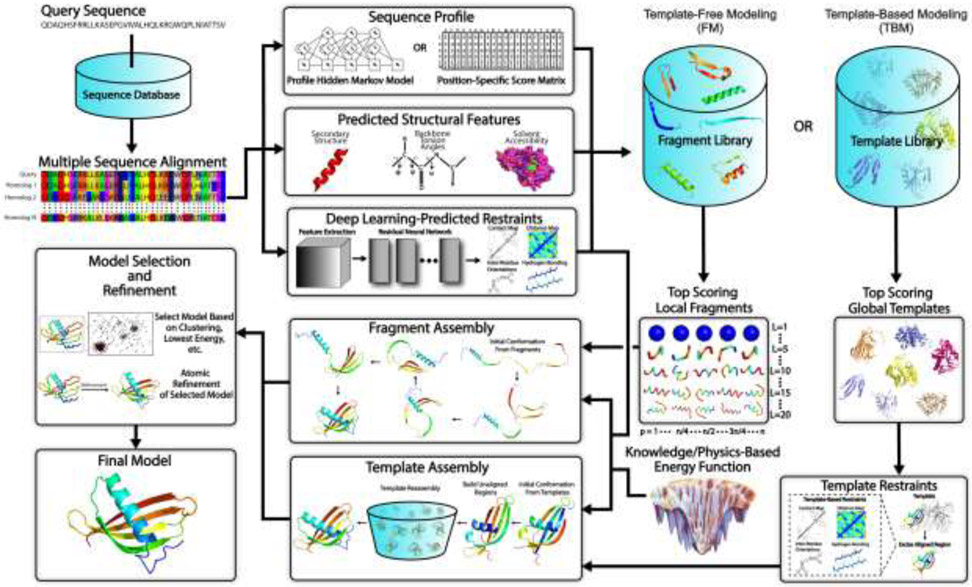
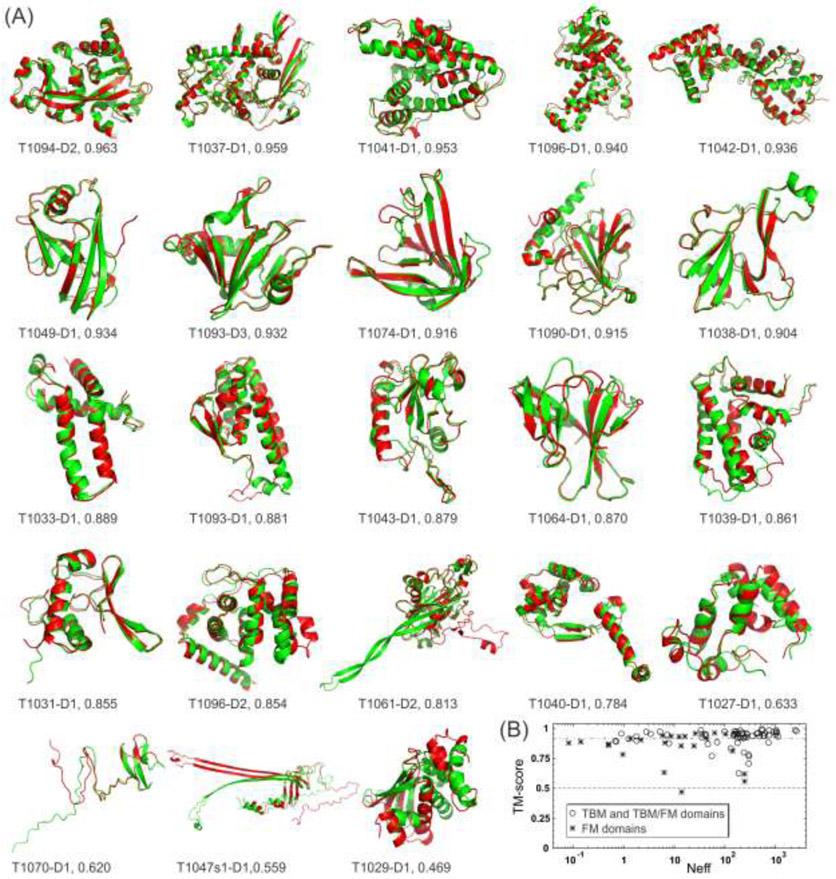
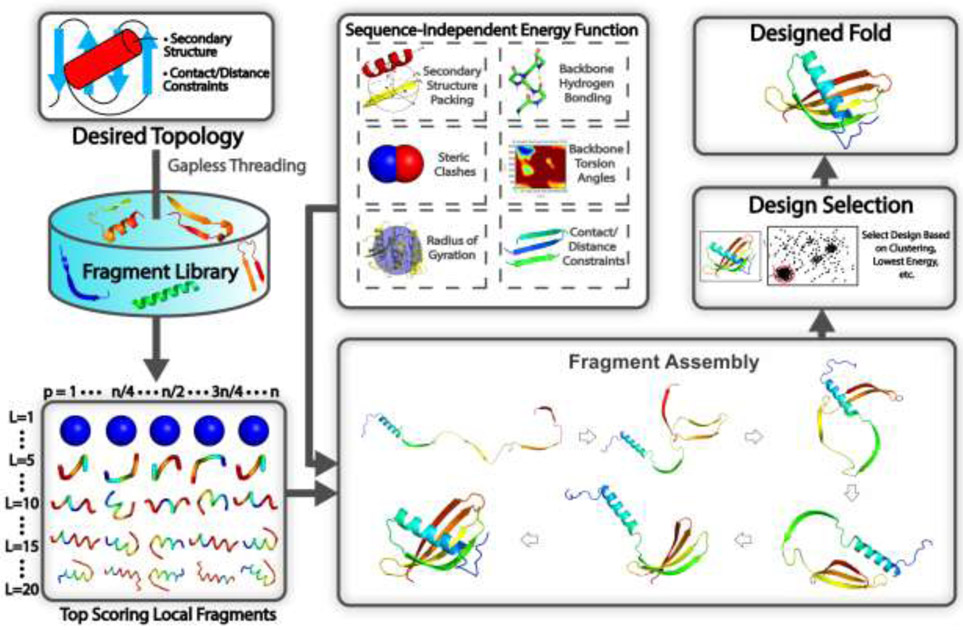
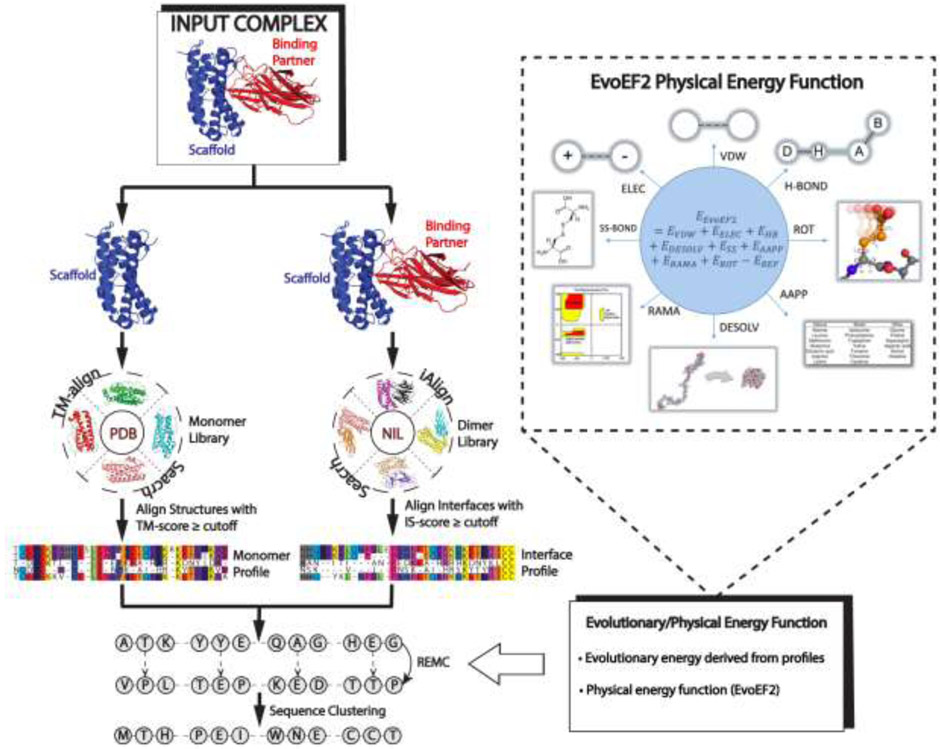
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
