

Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population
- PMID: 33639975
- PMCID: PMC7912818
- DOI: 10.1186/s13023-021-01729-0
Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population
Erratum in
-
Correction to: Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population.Orphanet J Rare Dis. 2021 Apr 1;16(1):154. doi: 10.1186/s13023-021-01786-5. Orphanet J Rare Dis. 2021. PMID: 33794951 Free PMC article. No abstract available.
Abstract
Background: X-linked hypophosphatemia (XLH) is a hereditary rare disease caused by loss-of-function mutations in PHEX gene leading tohypophosphatemia and high renal loss of phosphate. Rickets and growth retardation are the major manifestations of XLH in children, but there is a broad phenotypic variability. Few publications have reported large series of patients. Current data on the clinical spectrum of the disease, the correlation with the underlying gene mutations, and the long-term outcome of patients on conventional treatment are needed, particularly because of the recent availability of new specific medications to treat XLH.
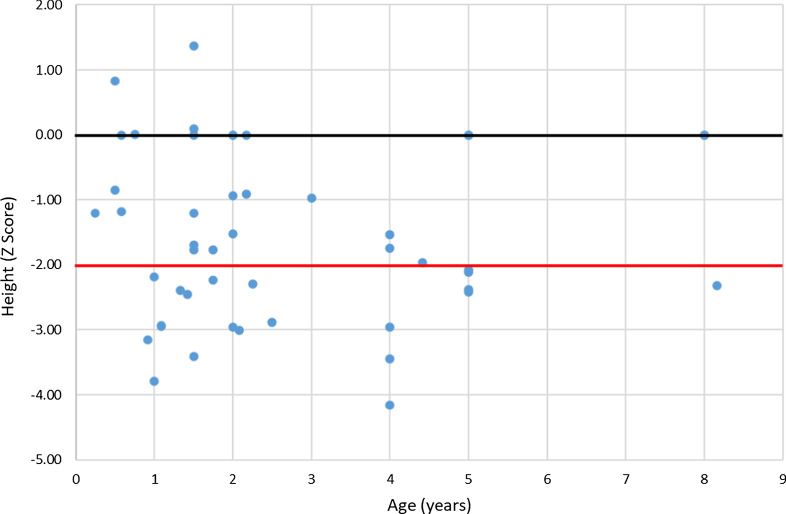
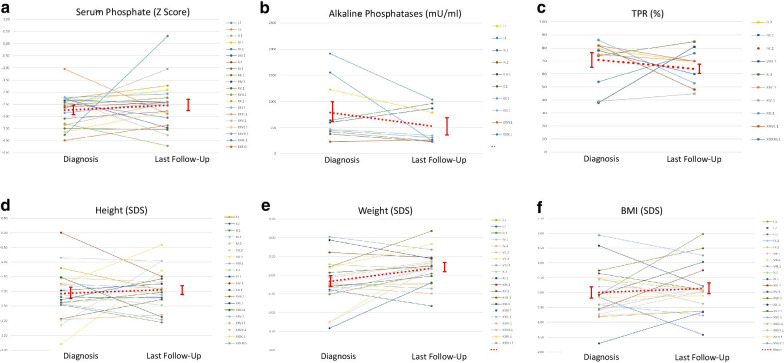
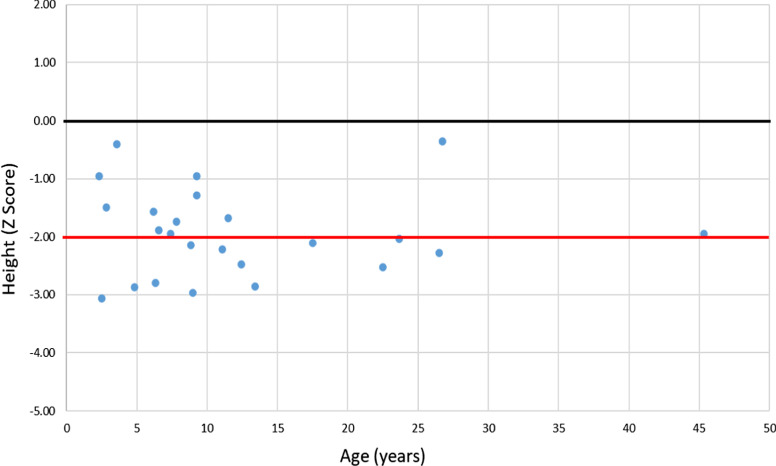
Results: The RenalTube database was used to retrospectively analyze 48 Spanish patients (15 men) from 39 different families, ranging from 3 months to 8 years and 2 months of age at the time of diagnosis (median age of 2.0 years), and with XLH confirmed by genetic analysis. Bone deformities, radiological signs of active rickets and growth retardation were the most common findings at diagnosis. Mean (± SEM) height was - 1.89 ± 0.19 SDS and 55% (22/40) of patients had height SDS below-2. All cases had hypophosphatemia, serum phosphate being - 2.81 ± 0.11 SDS. Clinical manifestations and severity of the disease were similar in both genders. No genotype-phenotype correlation was found. Conventional treatment did not attenuate growth retardation after a median follow up of 7.42 years (IQR = 11.26; n = 26 patients) and failed to normalize serum concentrations of phosphate. Eleven patients had mild hyperparathyroidism and 8 patients nephrocalcinosis.
Conclusions: This study shows that growth retardation and rickets were the most prevalent clinical manifestations at diagnosis in a large series of Spanish pediatric patients with XLH confirmed by mutations in the PHEX gene. Traditional treatment with phosphate and vitamin D supplements did not improve height or corrected hypophosphatemia and was associated with a risk of hyperparathyroidism and nephrocalcinosis. The severity of the disease was similar in males and females.
Keywords: Bone deformities; Growth retardation; Inherited hypophosphatemia; Rickets; XLH.
Conflict of interest statement
The study has been partially funded by Kyowa Kirin Farmacéutica S.L.U. This company produces the drug Crysvita® (burosumab). Nevertheless, no patients in this study had been or were being treated with burosumab at the time of data collection.
Figures
References
-
- Morey M, Castro-Feijóo L, Barreiro J, Cabanas P, Pombo M, Gil M, et al. Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: Tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type. BMC Med Genet. 2011;12(1):116. doi: 10.1186/1471-2350-12-116. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
