

Biallelic loss-of-function variants in PLD1 cause congenital right-sided cardiac valve defects and neonatal cardiomyopathy
- PMID: 33645542
- PMCID: PMC7919725
- DOI: 10.1172/JCI142148
Biallelic loss-of-function variants in PLD1 cause congenital right-sided cardiac valve defects and neonatal cardiomyopathy
Abstract
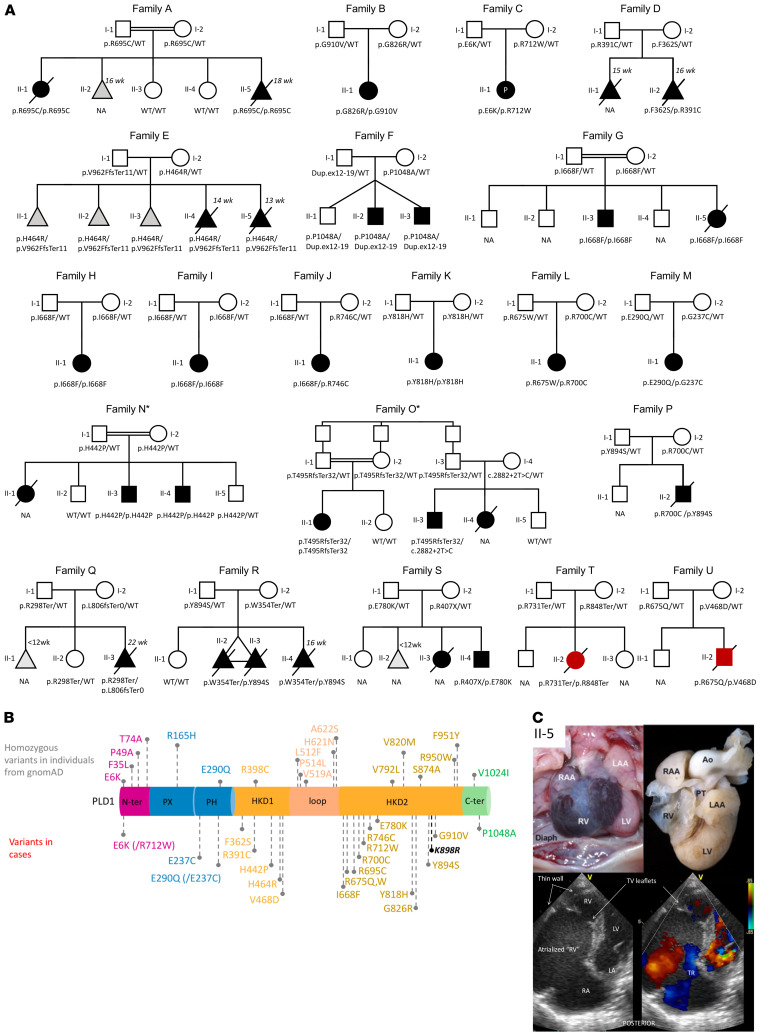
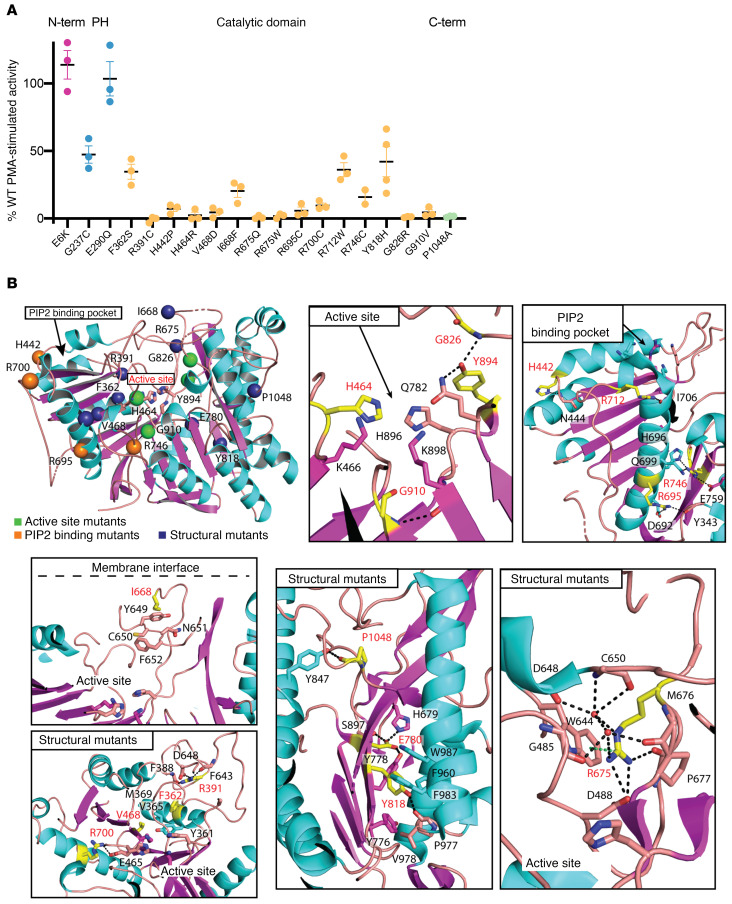
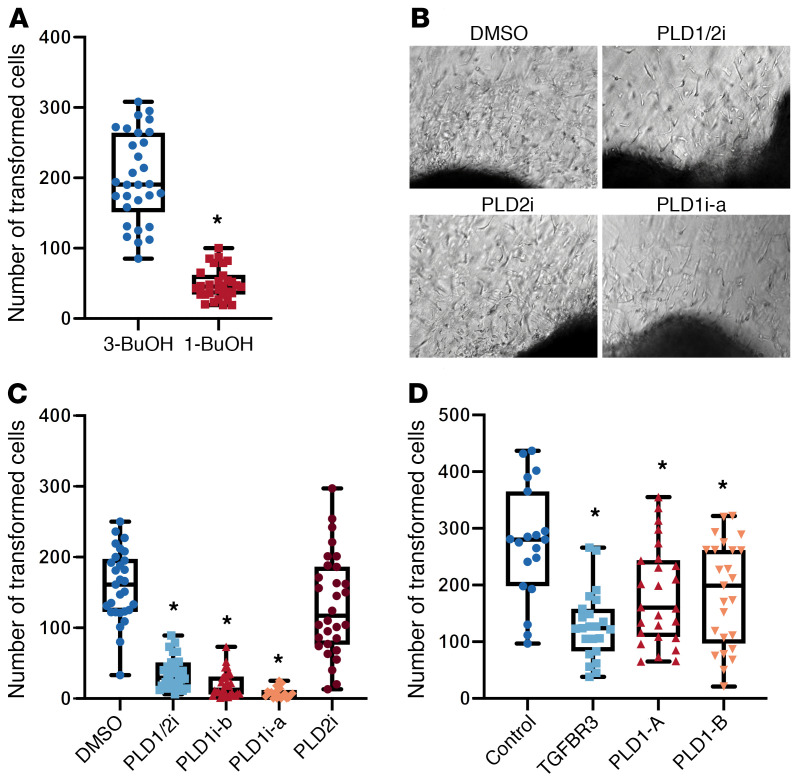
Congenital heart disease is the most common type of birth defect, accounting for one-third of all congenital anomalies. Using whole-exome sequencing of 2718 patients with congenital heart disease and a search in GeneMatcher, we identified 30 patients from 21 unrelated families of different ancestries with biallelic phospholipase D1 (PLD1) variants who presented predominantly with congenital cardiac valve defects. We also associated recessive PLD1 variants with isolated neonatal cardiomyopathy. Furthermore, we established that p.I668F is a founder variant among Ashkenazi Jews (allele frequency of ~2%) and describe the phenotypic spectrum of PLD1-associated congenital heart defects. PLD1 missense variants were overrepresented in regions of the protein critical for catalytic activity, and, correspondingly, we observed a strong reduction in enzymatic activity for most of the mutant proteins in an enzymatic assay. Finally, we demonstrate that PLD1 inhibition decreased endothelial-mesenchymal transition, an established pivotal early step in valvulogenesis. In conclusion, our study provides a more detailed understanding of disease mechanisms and phenotypic expression associated with PLD1 loss of function.
Trial registration: ClinicalTrials.gov NCT01196182.
Keywords: Cardiology; Cardiovascular disease; Genetic diseases; Genetics; Heart failure.
Conflict of interest statement
Figures
Similar articles
-
Congenital valvular defects associated with deleterious mutations in the PLD1 gene.J Med Genet. 2017 Apr;54(4):278-286. doi: 10.1136/jmedgenet-2016-104259. Epub 2016 Oct 31. J Med Genet. 2017. PMID: 27799408
-
Triple repeated fetal congenital heart disease linked to PLD1 mutation: a case report.J Med Case Rep. 2023 Sep 29;17(1):411. doi: 10.1186/s13256-023-04149-9. J Med Case Rep. 2023. PMID: 37770978 Free PMC article.
-
Prenatal detection of novel compound heterozygous variants of the PLD1 gene in a fetus with congenital heart disease.Front Genet. 2024 Nov 1;15:1498485. doi: 10.3389/fgene.2024.1498485. eCollection 2024. Front Genet. 2024. PMID: 39553471 Free PMC article.
-
Bicuspid Aortic Valve: a Review with Recommendations for Genetic Counseling.J Genet Couns. 2016 Dec;25(6):1171-1178. doi: 10.1007/s10897-016-0002-6. Epub 2016 Aug 22. J Genet Couns. 2016. PMID: 27550231 Free PMC article. Review.
-
Genetic Etiology of Left-Sided Obstructive Heart Lesions: A Story in Development.J Am Heart Assoc. 2021 Jan 19;10(2):e019006. doi: 10.1161/JAHA.120.019006. Epub 2021 Jan 12. J Am Heart Assoc. 2021. PMID: 33432820 Free PMC article. Review.
Cited by
-
Detection of Novel Pathogenic Variants in Two Families with Recurrent Fetal Congenital Heart Defects.Pharmgenomics Pers Med. 2023 Mar 8;16:173-181. doi: 10.2147/PGPM.S394120. eCollection 2023. Pharmgenomics Pers Med. 2023. PMID: 36923242 Free PMC article.
-
Structure and regulation of human phospholipase D.Adv Biol Regul. 2021 Jan;79:100783. doi: 10.1016/j.jbior.2020.100783. Epub 2021 Jan 3. Adv Biol Regul. 2021. PMID: 33495125 Free PMC article. Review.
-
Defining the proximal interaction networks of Arf GTPases reveals a mechanism for the regulation of PLD1 and PI4KB.EMBO J. 2022 Sep 1;41(17):e110698. doi: 10.15252/embj.2022110698. Epub 2022 Jul 17. EMBO J. 2022. PMID: 35844135 Free PMC article.
-
Simultaneous CNV-seq and WES: An effective strategy for molecular diagnosis of unexplained fetal structural anomalies.Heliyon. 2024 Oct 15;10(20):e39392. doi: 10.1016/j.heliyon.2024.e39392. eCollection 2024 Oct 30. Heliyon. 2024. PMID: 39502218 Free PMC article.
-
Somatic GATA4 mutation contributes to tetralogy of Fallot.Exp Ther Med. 2024 Jan 8;27(2):91. doi: 10.3892/etm.2024.12379. eCollection 2024 Feb. Exp Ther Med. 2024. PMID: 38274337 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
