

Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens
- PMID: 33649593
- PMCID: PMC8011839
- DOI: 10.1038/s41588-021-00778-2
Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens
Abstract
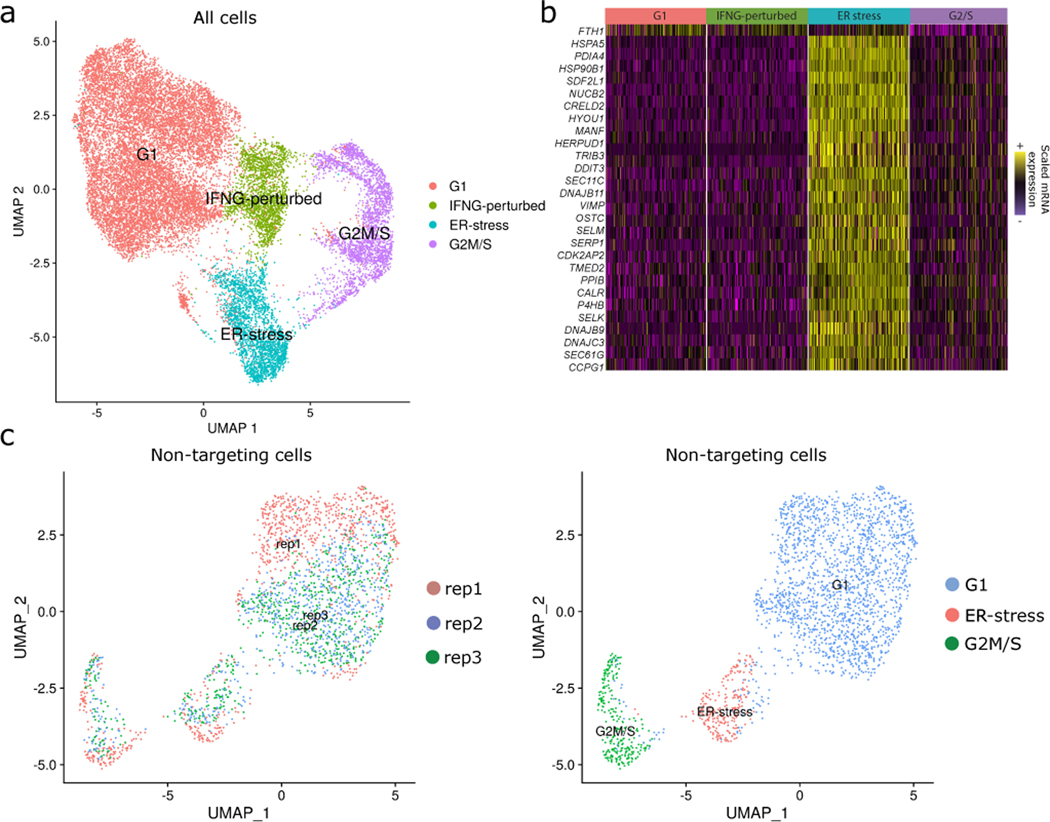
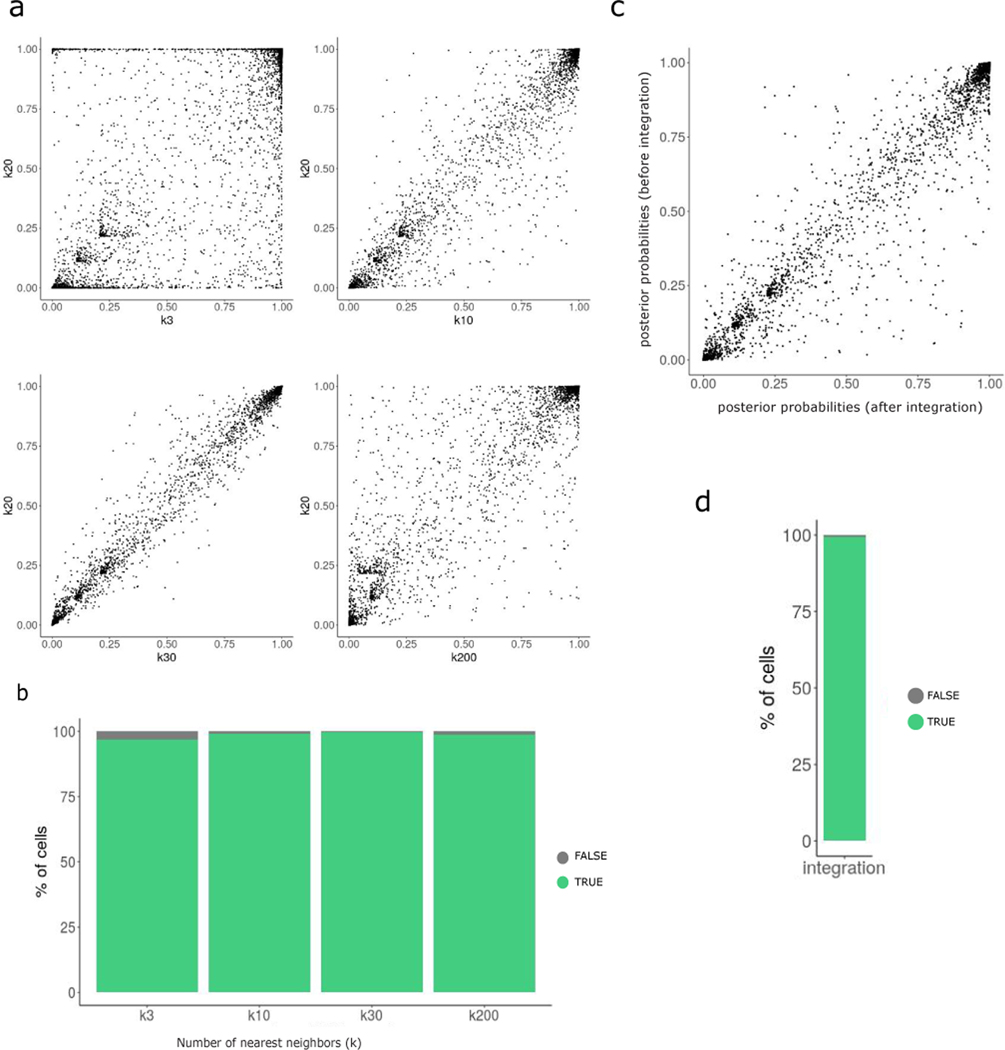
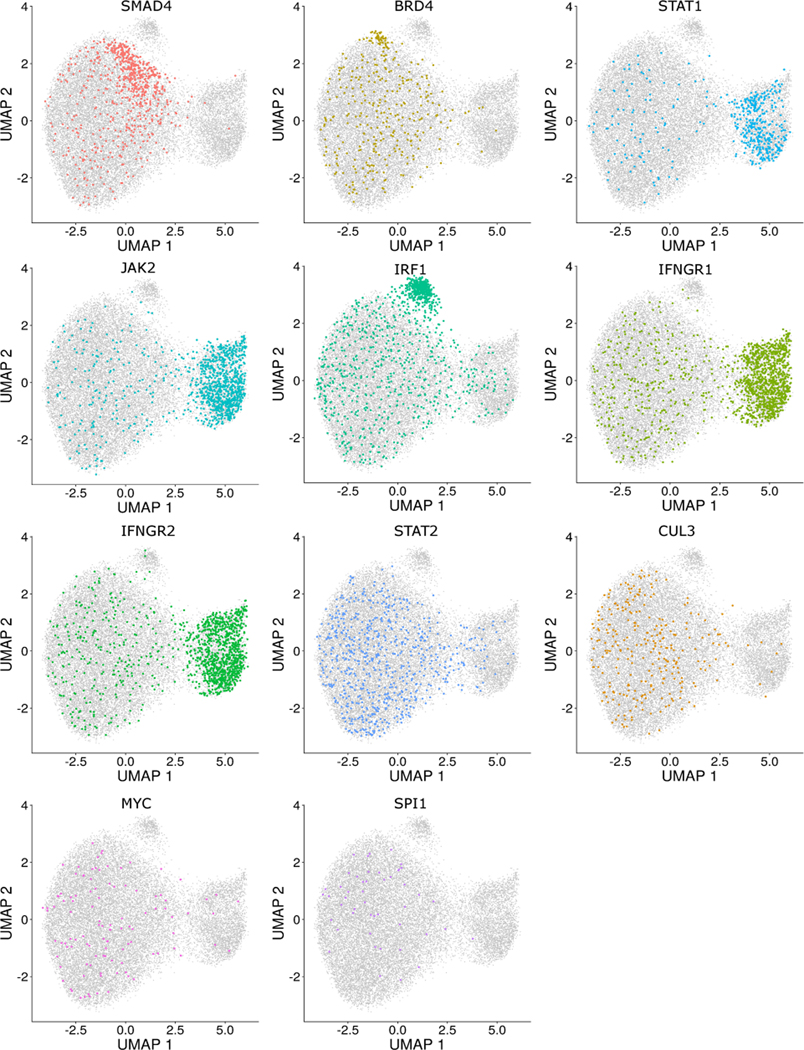
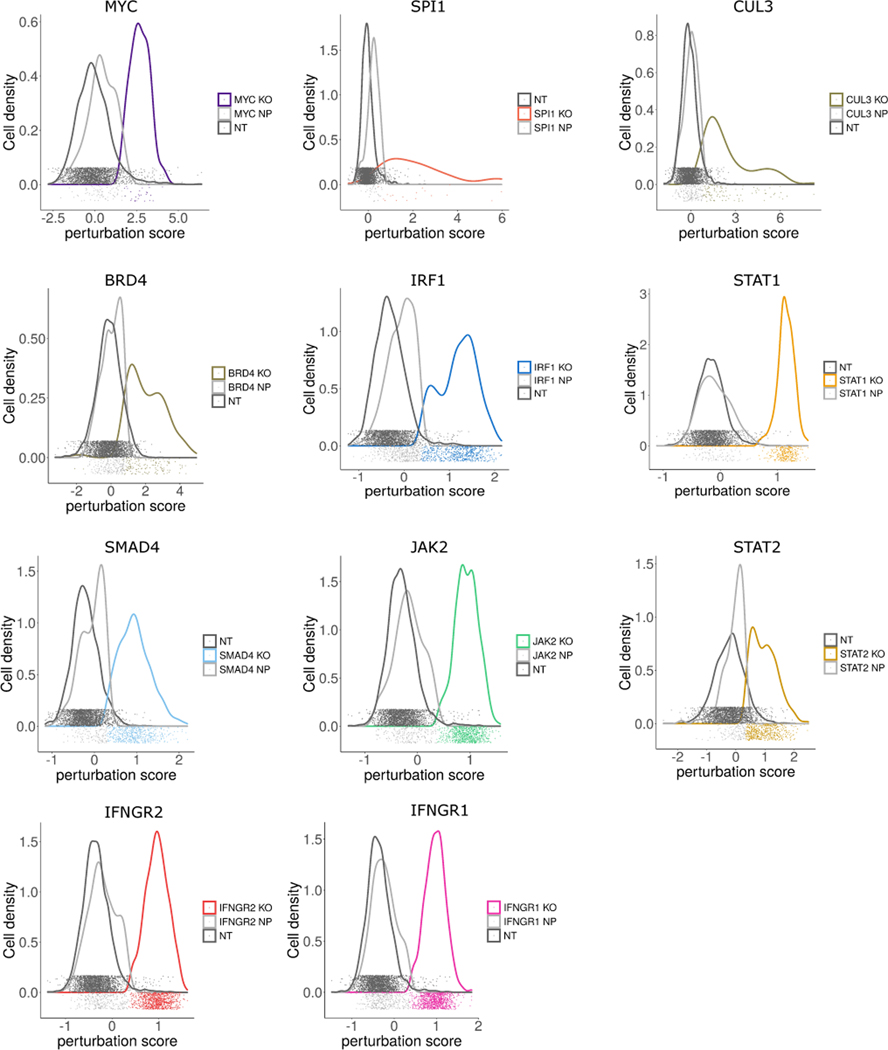
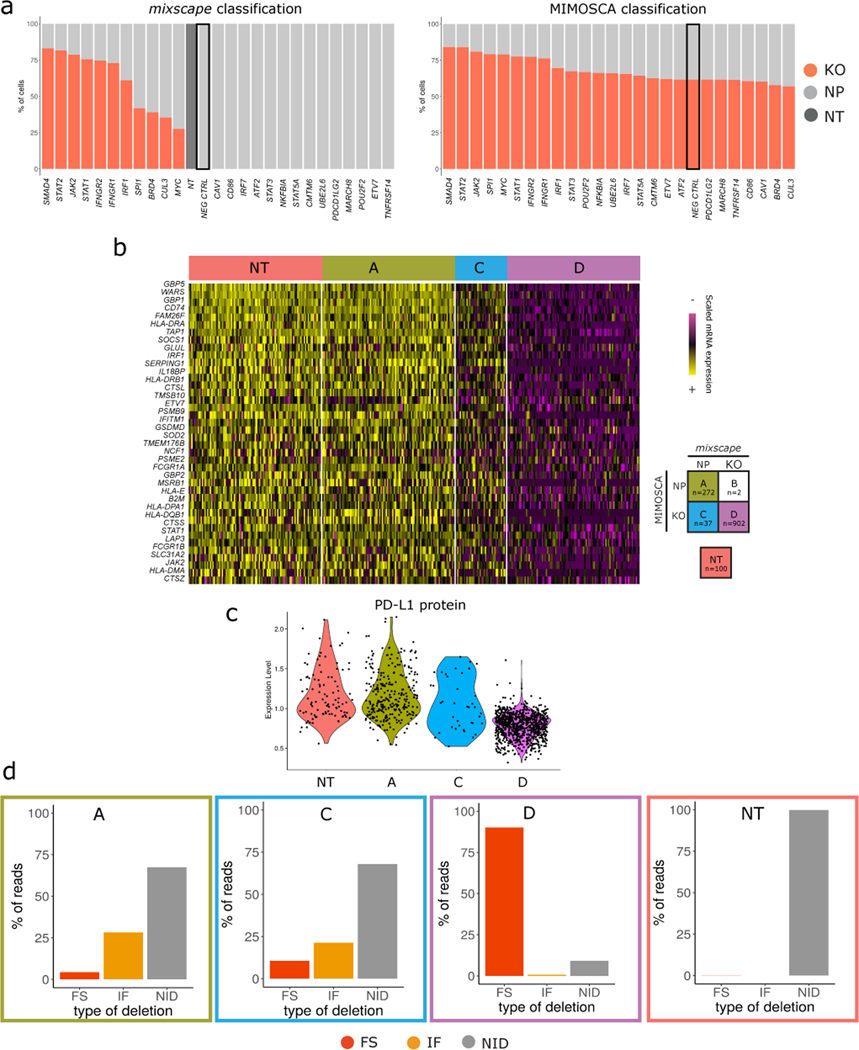
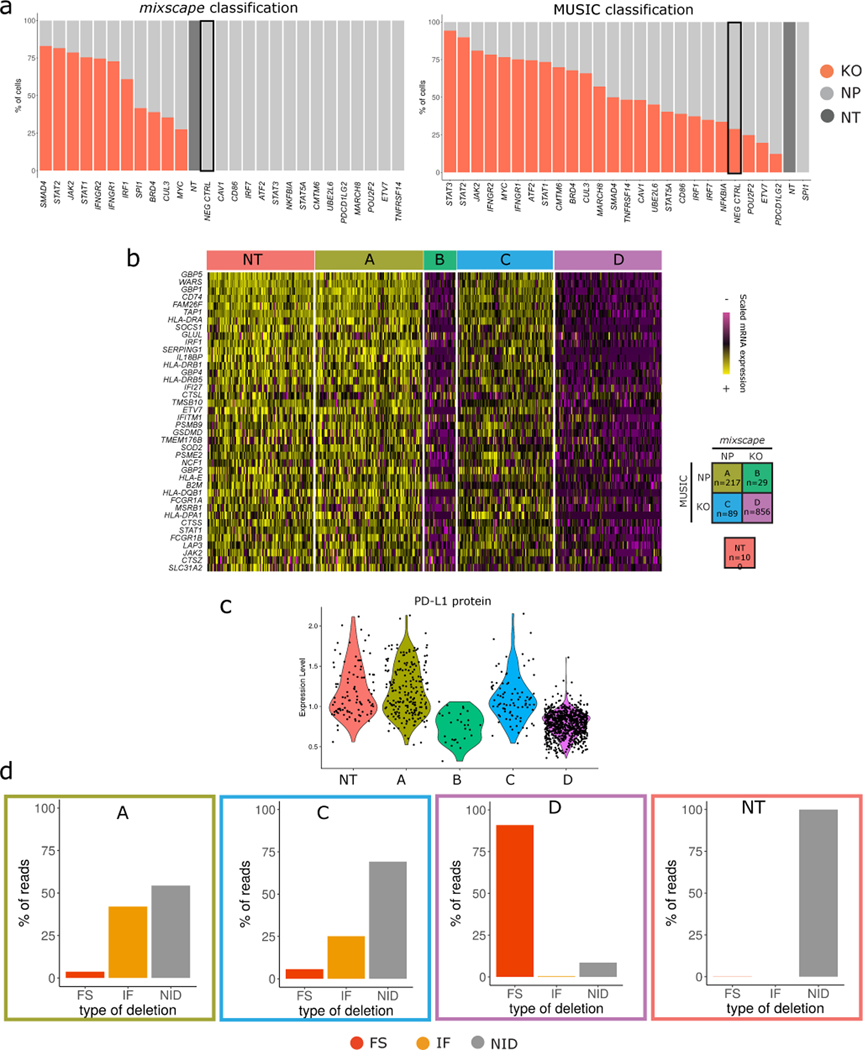
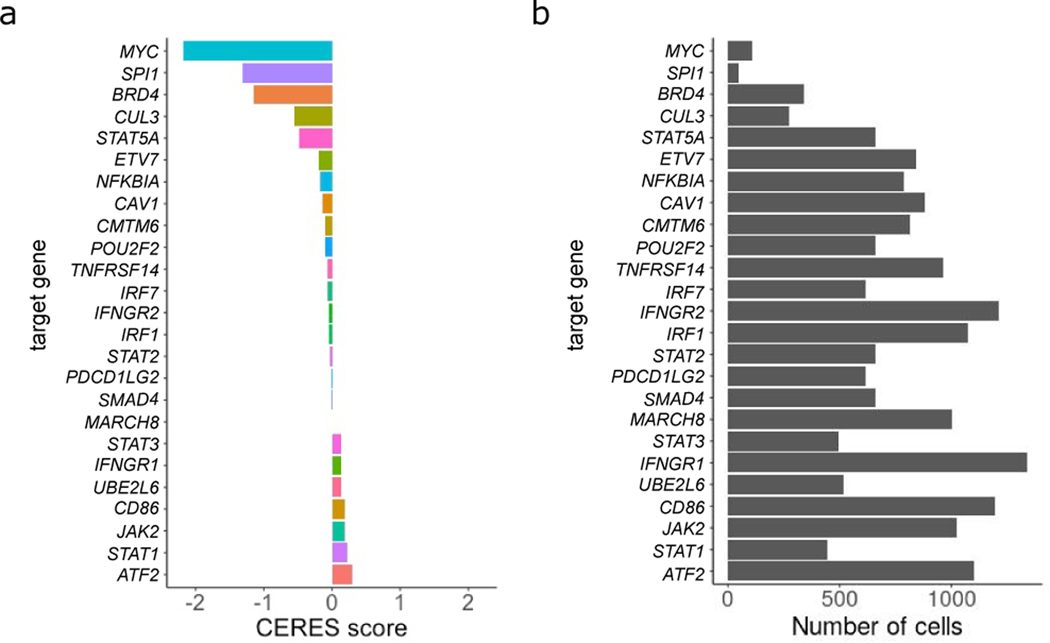
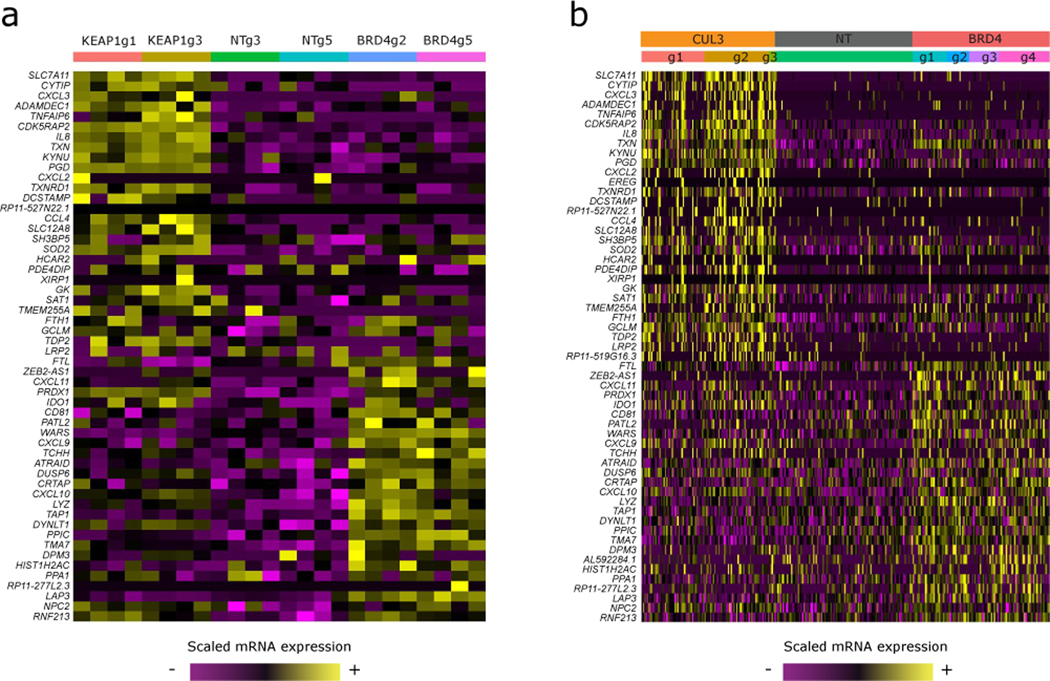
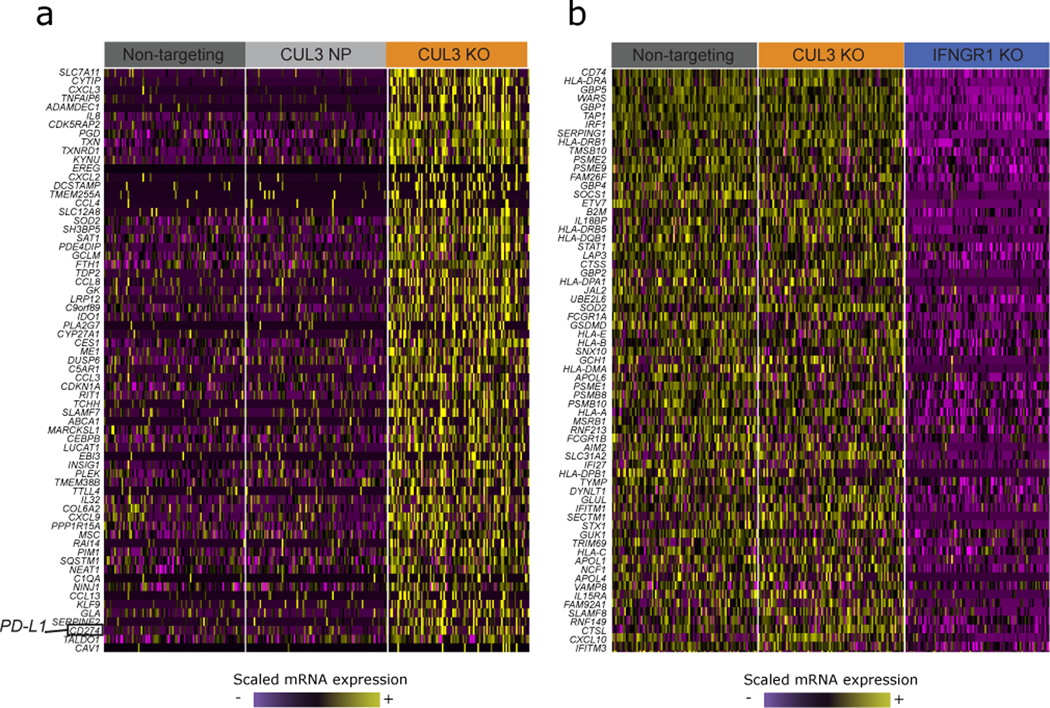
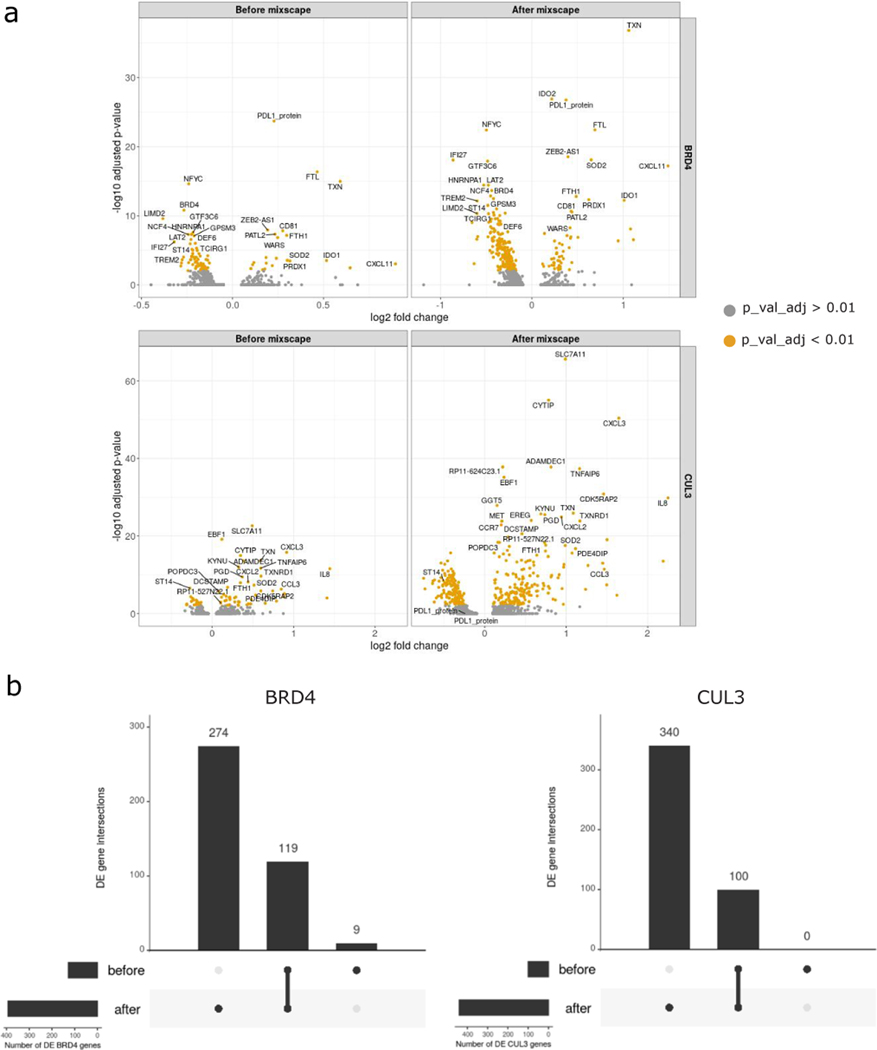
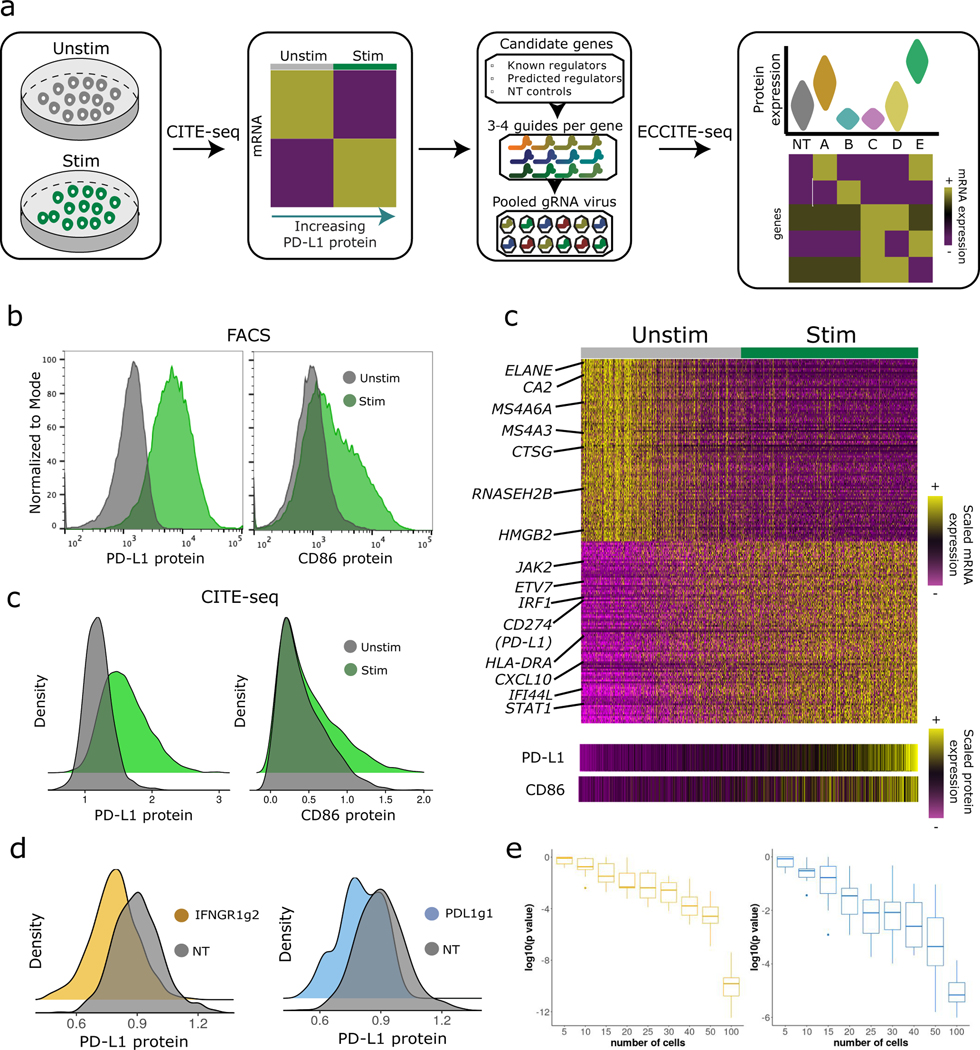
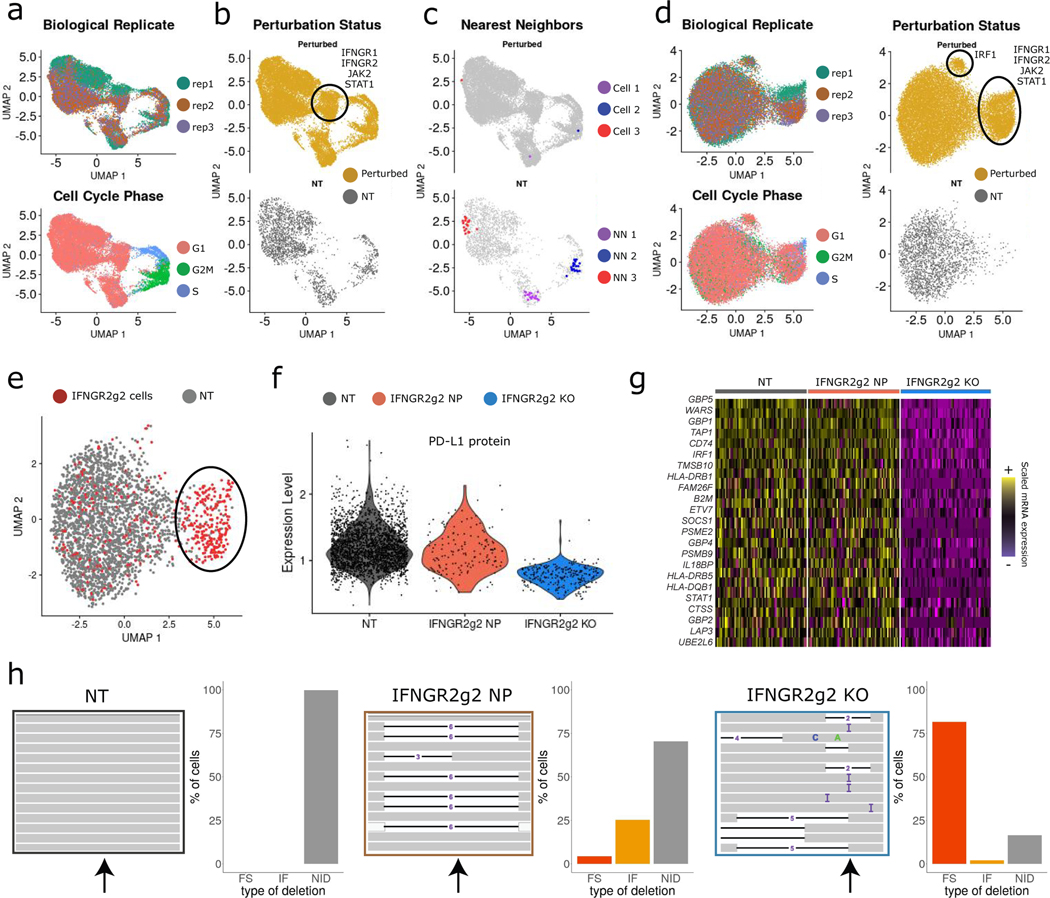
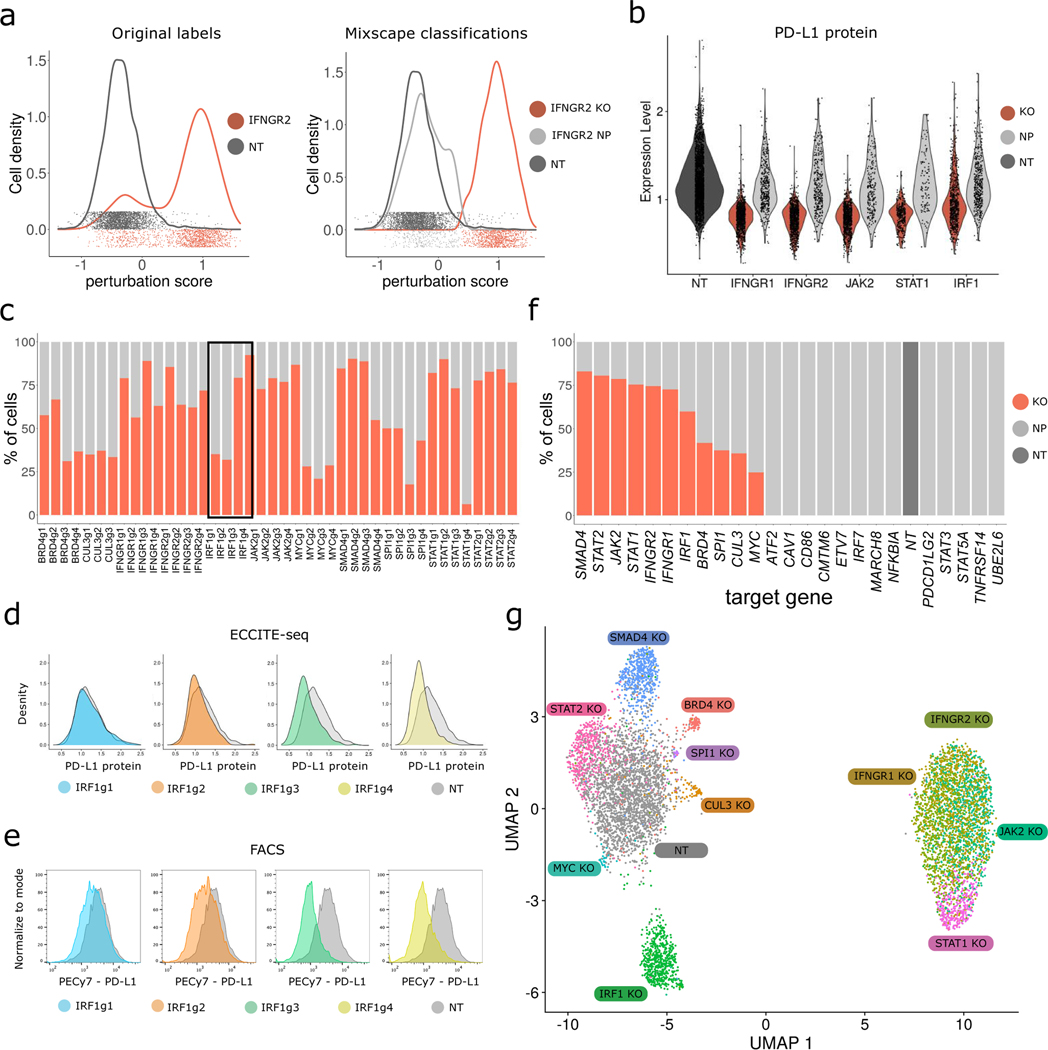
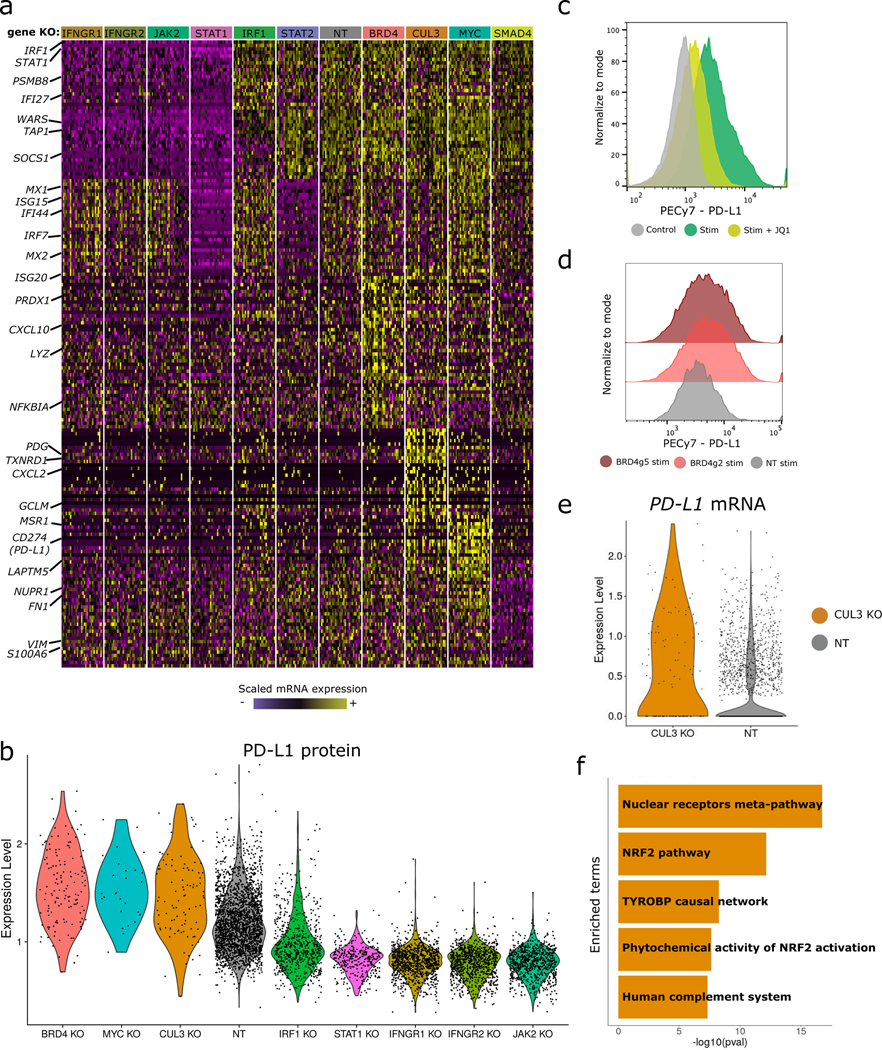
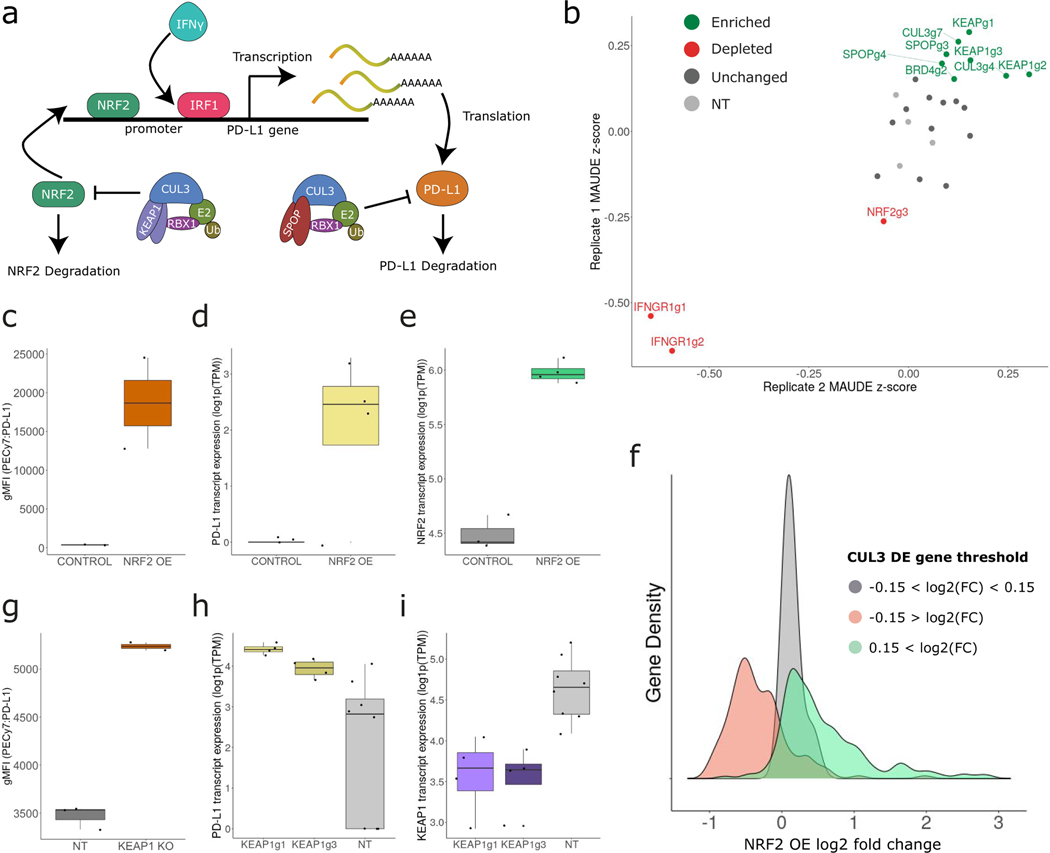
The expression of inhibitory immune checkpoint molecules, such as programmed death-ligand (PD-L)1, is frequently observed in human cancers and can lead to the suppression of T cell-mediated immune responses. Here, we apply expanded CRISPR-compatible (EC)CITE-seq, a technology that combines pooled CRISPR screens with single-cell mRNA and surface protein measurements, to explore the molecular networks that regulate PD-L1 expression. We also develop a computational framework, mixscape, that substantially improves the signal-to-noise ratio in single-cell perturbation screens by identifying and removing confounding sources of variation. Applying these tools, we identify and validate regulators of PD-L1 and leverage our multimodal data to identify both transcriptional and post-transcriptional modes of regulation. Specifically, we discover that the Kelch-like protein KEAP1 and the transcriptional activator NRF2 mediate the upregulation of PD-L1 after interferon (IFN)-γ stimulation. Our results identify a new mechanism for the regulation of immune checkpoints and present a powerful analytical framework for the analysis of multimodal single-cell perturbation screens.
Figures
References
METHODS-ONLY REFERENCES
-
- Ashland OR: Becton, Dickinson and Company. FlowJo TM Software, Version 10.6.2. 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
