

Composite phaeochromocytomas-a systematic review of published literature
- PMID: 33651160
- PMCID: PMC8933353
- DOI: 10.1007/s00423-021-02129-5
Composite phaeochromocytomas-a systematic review of published literature
Abstract
Introduction: Composite phaeochromocytoma is a tumour containing a separate tumour of neuronal origin in addition to a chromaffin cell tumour. This study reports on two cases from a single centre's records and presents a systematic literature review of composite phaeochromocytomas.
Methods: In addition to describing 2 case reports, a systematic search of the Medline database from inception up to April 2020 was done for human case reports on composite phaeochromocytomas. Relevant titles and/or abstracts were screened, and full texts were reviewed to identify appropriate studies. Data was extracted and a descriptive analysis of presentation, clinical features, management strategies and outcomes was performed. The quality of included studies was assessed using a critical appraisal checklist.
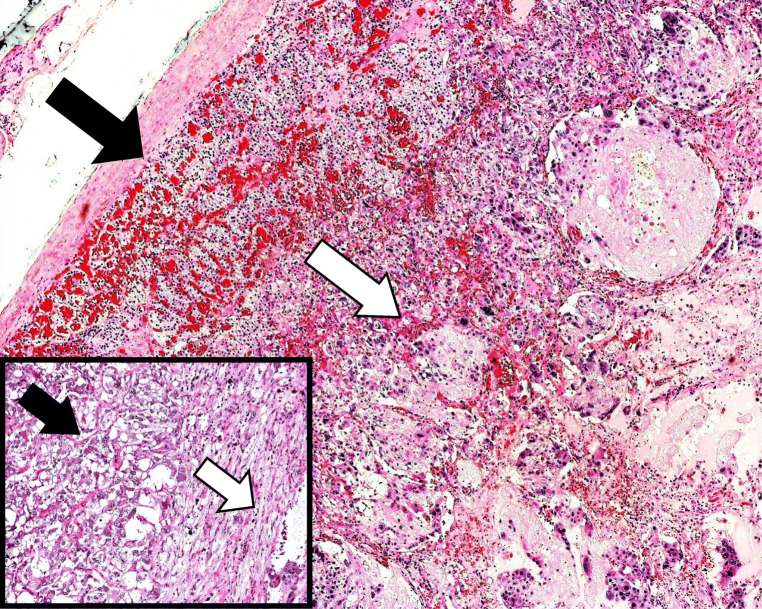
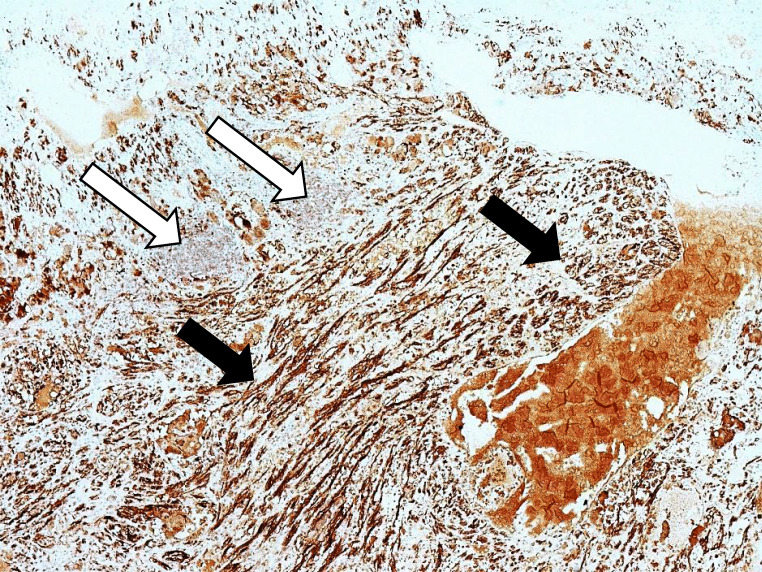
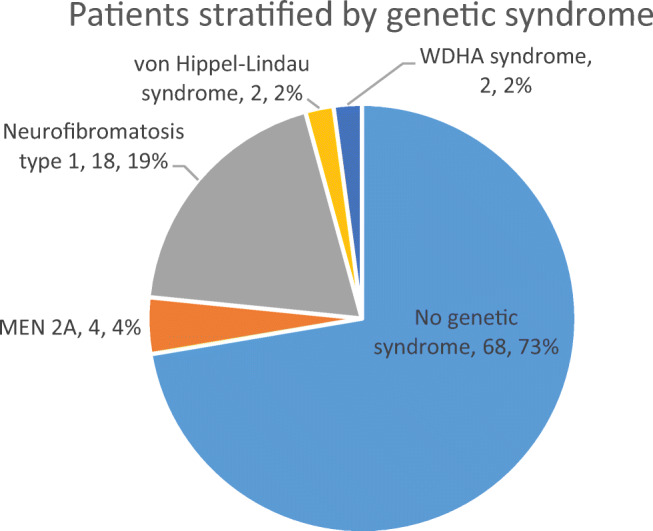
Results: There were 62 studies included, with a total of 94 patients. Of 91 patients where data was available, the median (range) age of patients was 48 (4-86) years. Of 90 patients where information was provided, 57% were female. In at least 28% of patients, a genetic cause was identified. Common presenting features include abdominal pain, palpable mass, cardiovascular and gastrointestinal symptoms. The most common tumour component with phaeochromocytoma is ganglioneuroma; other components include ganglioneuroblastoma, neuroblastoma and malignant peripheral nerve sheath tumours. In patients with follow-up data (n=48), 85% of patients were alive and well at a median (range) follow-up time of 18 (0.5-168) months.
Conclusion: Composite phaeochromocytoma is a rare tumour, with a significant genetic predisposition. This review summarises available epidemiological data, which will be useful for clinicians managing this rare condition.
Keywords: Adrenal; Composite tumours; Ganglioneuroblastoma; Ganglioneuroma; Incidentaloma; Neuroblastoma; Phaeochromocytoma; Schwannoma.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Manea M, Marcu D, Bratu O, et al. Pheochromocytoma – clinical manifestations, diagnosis and current perioperative management. J Mind Med Sci. 2019;6:243–247. doi: 10.22543/7674.62.P243247. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
