

Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment
- PMID: 33651880
- PMCID: PMC7933992
- DOI: 10.1084/jem.20200920
Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment
Abstract
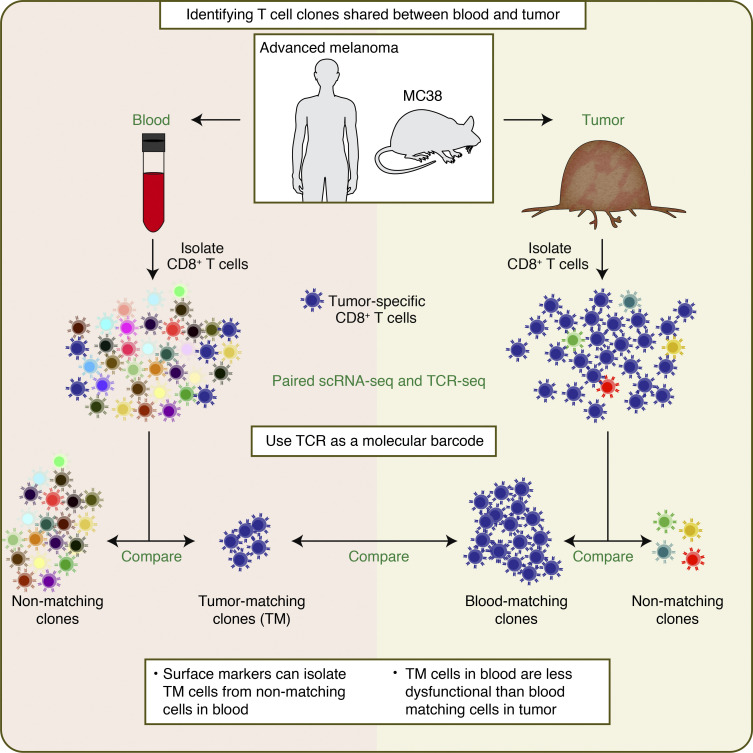
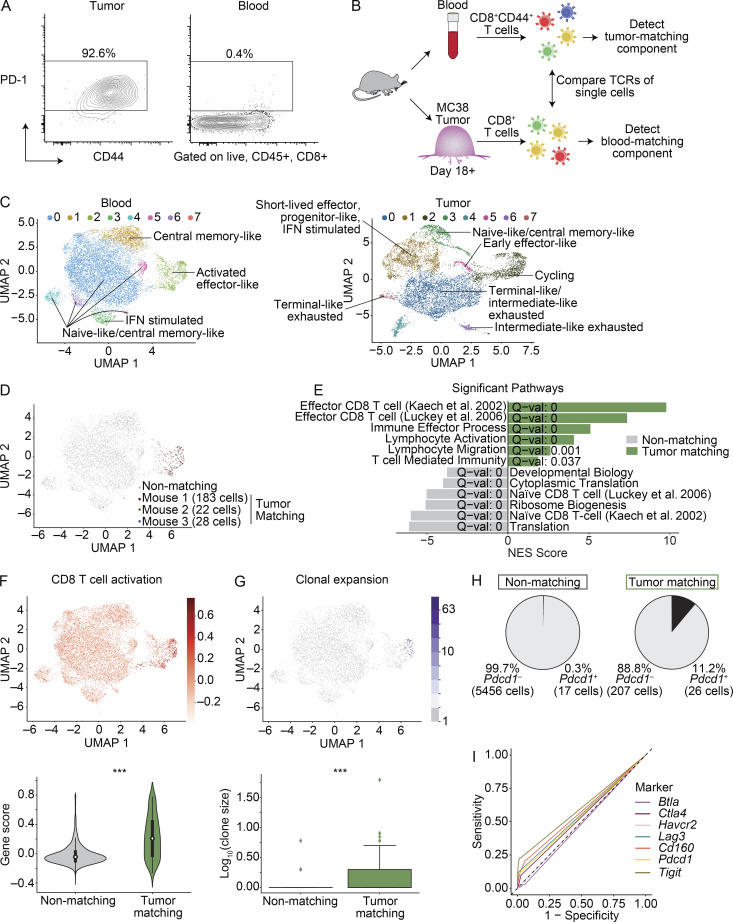
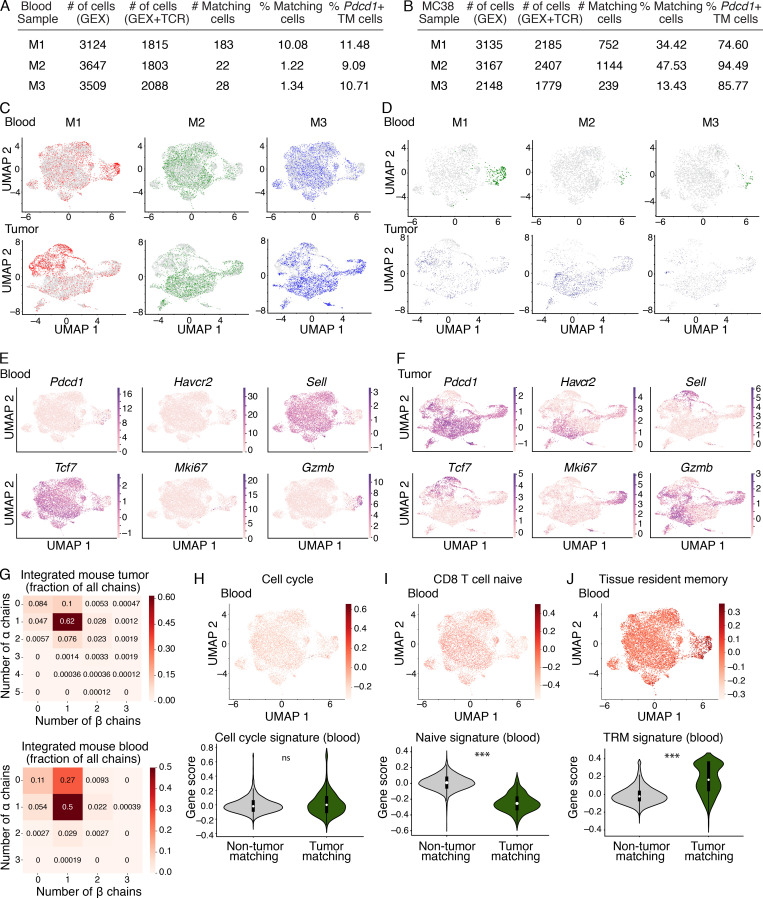
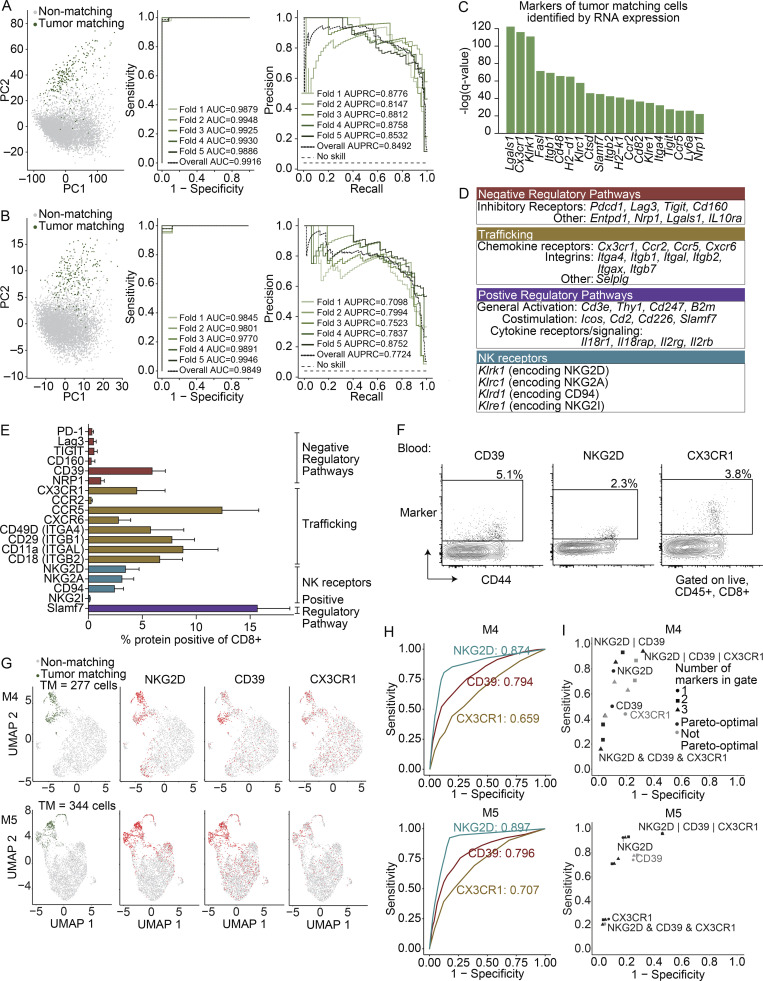
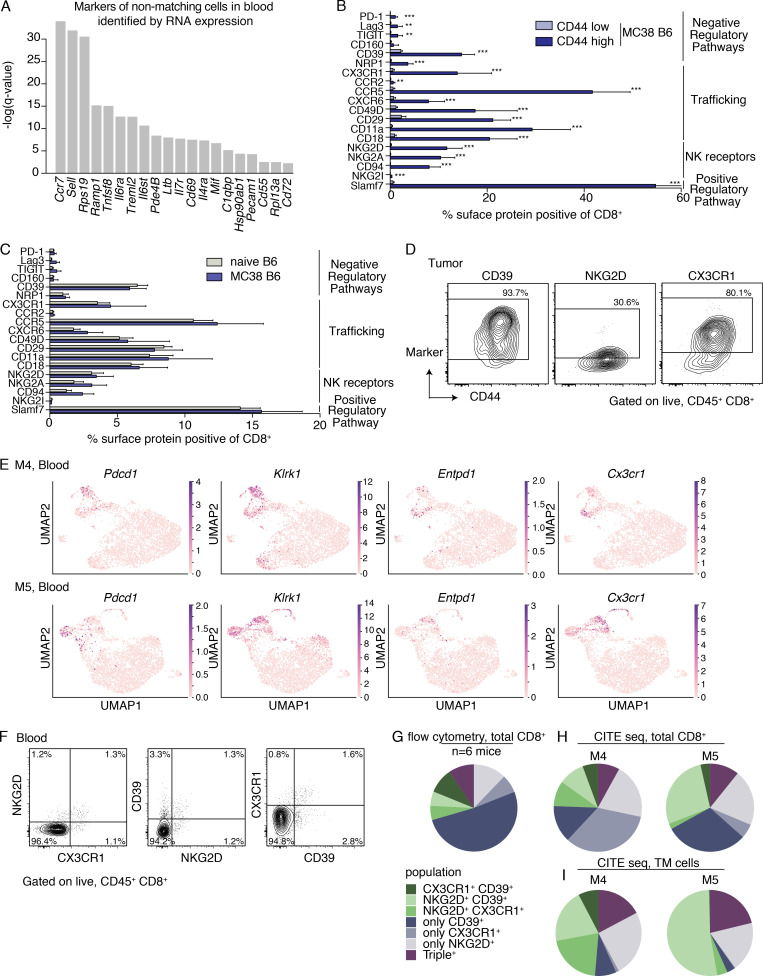
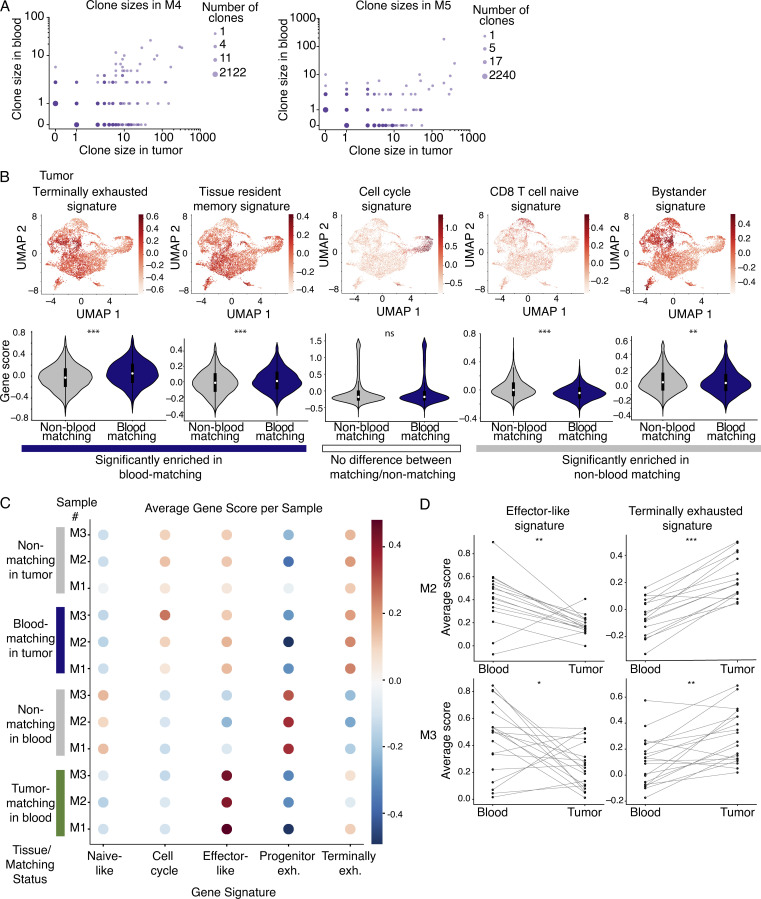
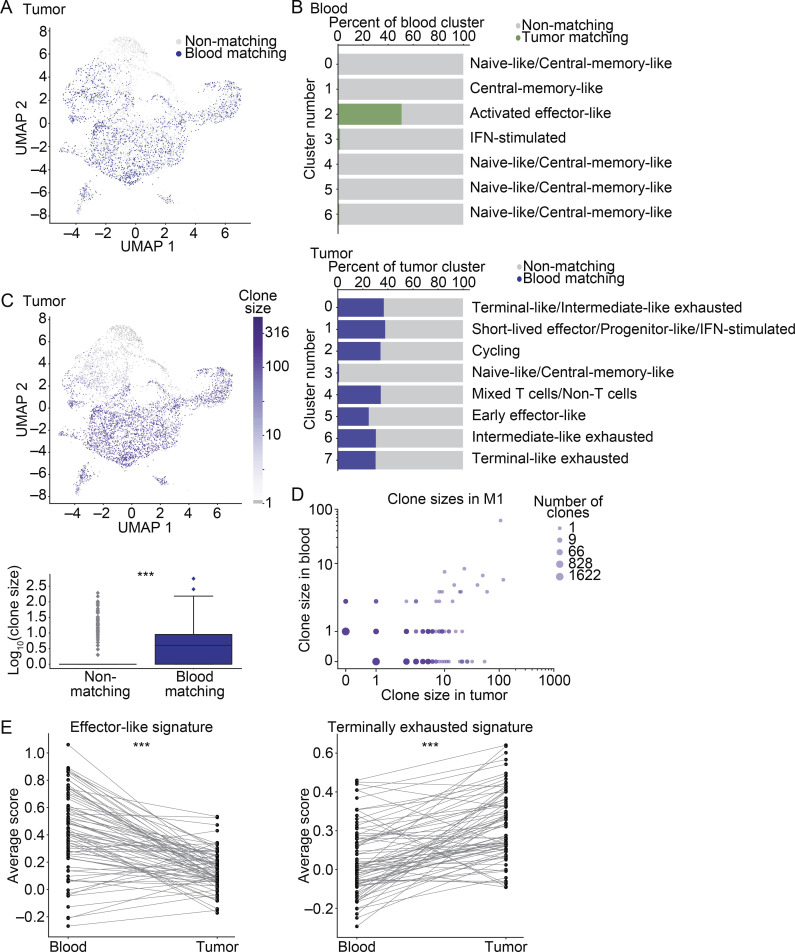
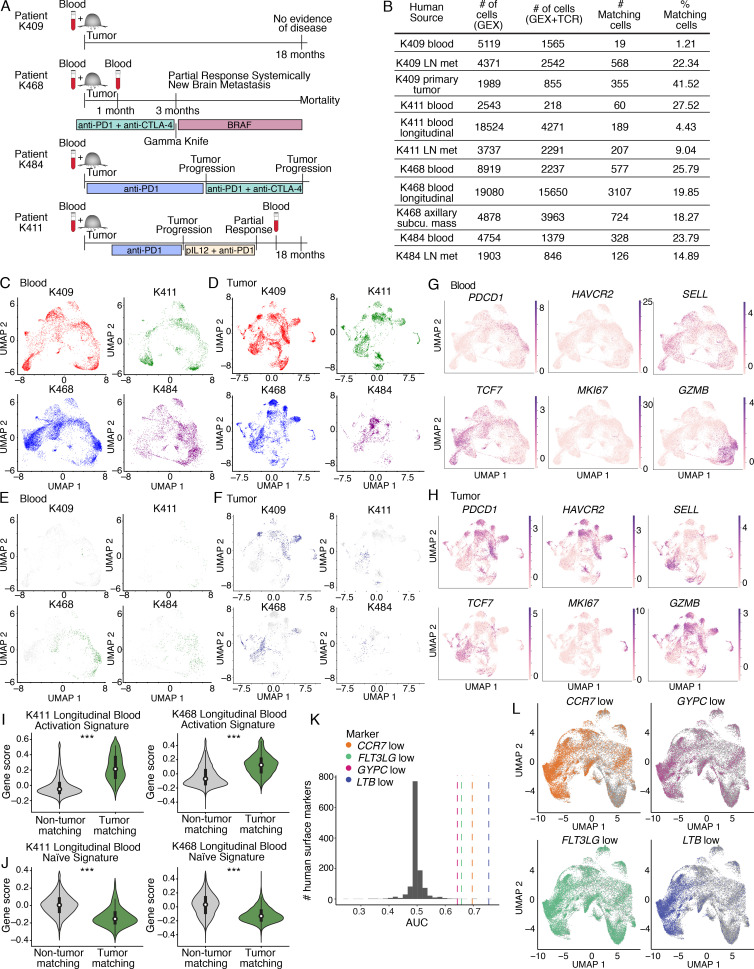
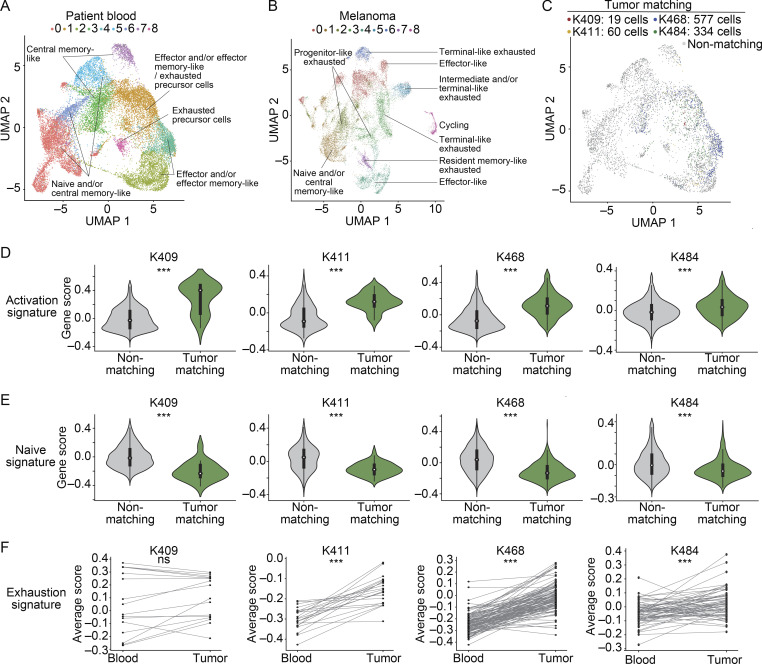
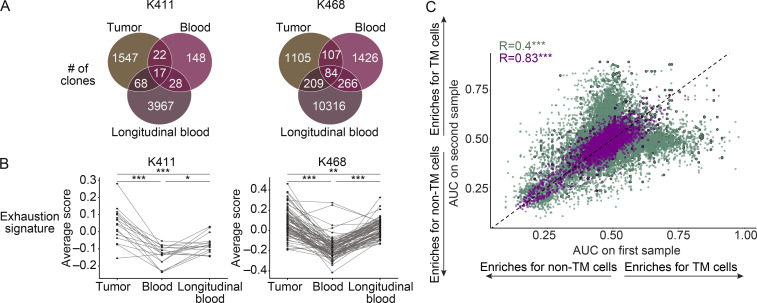
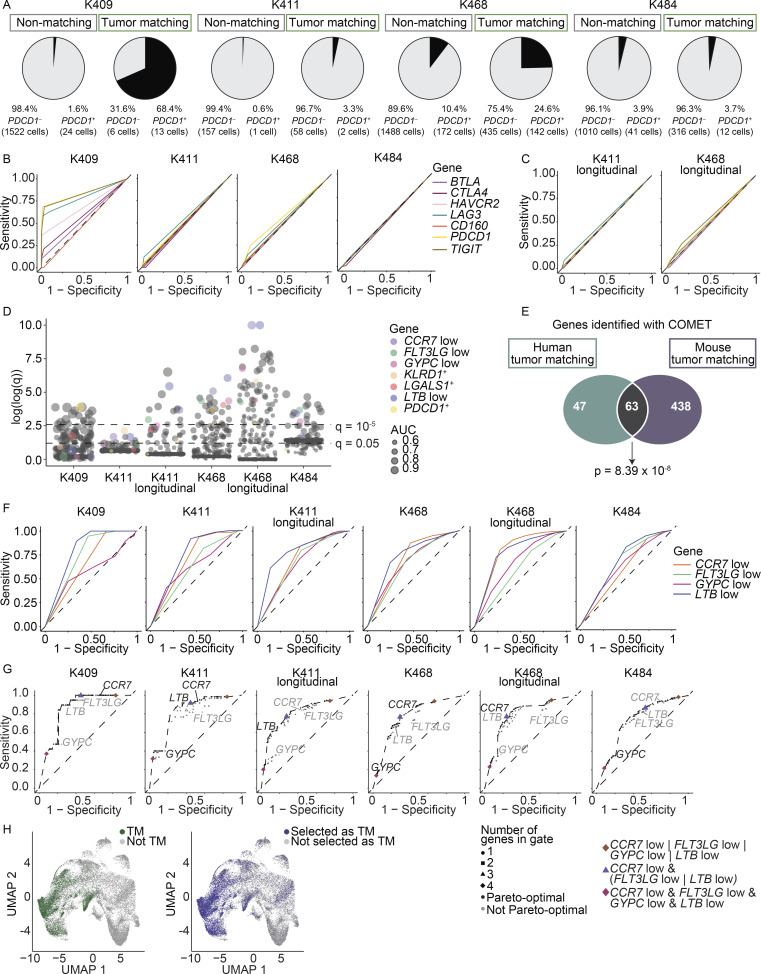
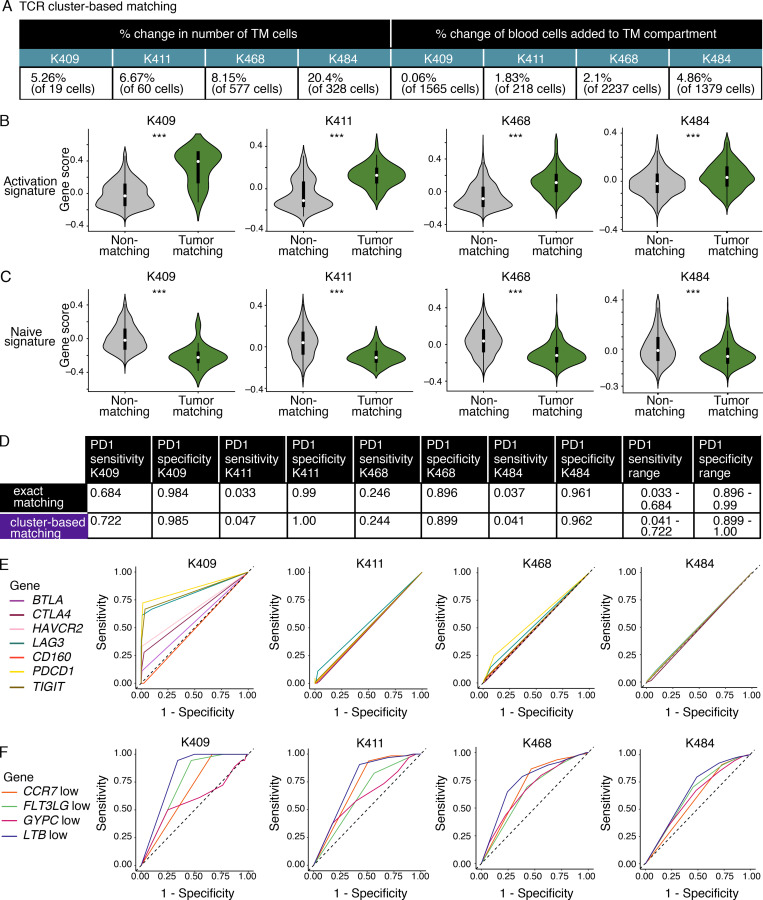
The ability to monitor anti-tumor CD8+ T cell responses in the blood has tremendous therapeutic potential. Here, we used paired single-cell RNA and TCR sequencing to detect and characterize "tumor-matching" (TM) CD8+ T cells in the blood of mice with MC38 tumors or melanoma patients using the TCR as a molecular barcode. TM cells showed increased activation compared with nonmatching T cells in blood and were less exhausted than matching cells in tumors. Importantly, PD-1, which has been used to identify putative circulating anti-tumor CD8+ T cells, showed poor sensitivity for identifying TM cells. By leveraging the transcriptome, we identified candidate cell surface markers for TM cells in mice and patients and validated NKG2D, CD39, and CX3CR1 in mice. These data show that the TCR can be used to identify tumor-relevant cells for characterization, reveal unique transcriptional properties of TM cells, and develop marker panels for tracking and analysis of these cells.
© 2021 Pauken et al.
Conflict of interest statement
Disclosures: K. Tsai reported personal fees from Regeneron, grants from Array/Pfizer, grants from Replimune, and grants from Oncosec outside the submitted work. M. Rosenblum reported other from TRex Bio and other from Sitryx Bio outside the submitted work. A. Daud reported grants from Merck, grants from BMS, other from Trex, grants from Pfizer, grants from Incyte, grants from Xencor, grants from Roche, and grants from Novartis during the conduct of the study as well as grants from Oncosec outside the submitted work. A. Sharpe reported grants from NIH R01 CA229851, grants from NIH U54 CA224088, grants from NIH P01 56299, and grants from NIH P01 39671 during the conduct of the study; personal fees from Surface Oncology, personal fees from Sqz Biotech, personal fees from Selecta, personal fees from Monopteros, personal fees from Elstar, personal fees from Elpiscience, grants from Novartis, grants from Roche, grants from Merck, grants from Ipsen, grants from UCB, and grants from Quark Ventures outside the submitted work. In addition, A. Sharpe had a patent number 7,432,059 with royalties paid (Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Leica, Mayo Clinic, Dako and Novartis), a patent number 7,722,868 with royalties paid (Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Leica, Mayo Clinic, Dako and Novartis), a patent number 8,652,465 licensed (Roche), a patent number 9,457,080 licensed (Roche), a patent number 9,683,048 licensed (Novartis), a patent number 9,815,898 licensed (Novartis), a patent number 9,845,356 licensed (Novartis), a patent number 10,202,454 licensed (Novartis), a patent number 10,457,733 licensed (Novartis), a patent number 9,580,684 issued (none), a patent number 9,988,452 issued (none), and a patent number 10,370,446 issued (none); A. Sharpe is on the scientific advisory boards for the Massachusetts General Cancer Center, Program in Cellular and Molecular Medicine at Boston Children's Hospital, and the Human Oncology and Pathogenesis Program at Memorial Sloan Kettering Cancer Center and is a scientific editor for the Journal of Experimental Medicine. M. Singer reported personal fees from Guardant Health outside the submitted work. No other disclosures were reported.
Figures
References
-
- Algazi, A., Bhatia S., Agarwala S., Molina M., Lewis K., Faries M., Fong L., Levine L.P., Franco M., Oglesby A., et al. . 2020. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol. 31(4):532–540. 10.1016/j.annonc.2019.12.008 - DOI - PubMed
-
- Beltra, J.C., Manne S., Abdel-Hakeem M.S., Kurachi M., Giles J.R., Chen Z., Casella V., Ngiow S.F., Khan O., Huang Y.J., et al. . 2020. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity. 52:825–841.e8. 10.1016/j.immuni.2020.04.014 - DOI - PMC - PubMed
-
- Best, J.A., Blair D.A., Knell J., Yang E., Mayya V., Doedens A., Dustin M.L., and Goldrath A.W.. Immunological Genome Project Consortium . 2013. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 14:404–412. 10.1038/ni.2536 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
