

vcf2fhir: a utility to convert VCF files into HL7 FHIR format for genomics-EHR integration
- PMID: 33653260
- PMCID: PMC7923512
- DOI: 10.1186/s12859-021-04039-1
vcf2fhir: a utility to convert VCF files into HL7 FHIR format for genomics-EHR integration
Abstract
Background: VCF formatted files are the lingua franca of next-generation sequencing, whereas HL7 FHIR is emerging as a standard language for electronic health record interoperability. A growing number of FHIR-based clinical genomics applications are emerging. Here, we describe an open source utility for converting variants from VCF format into HL7 FHIR format.
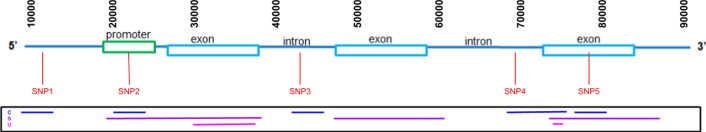
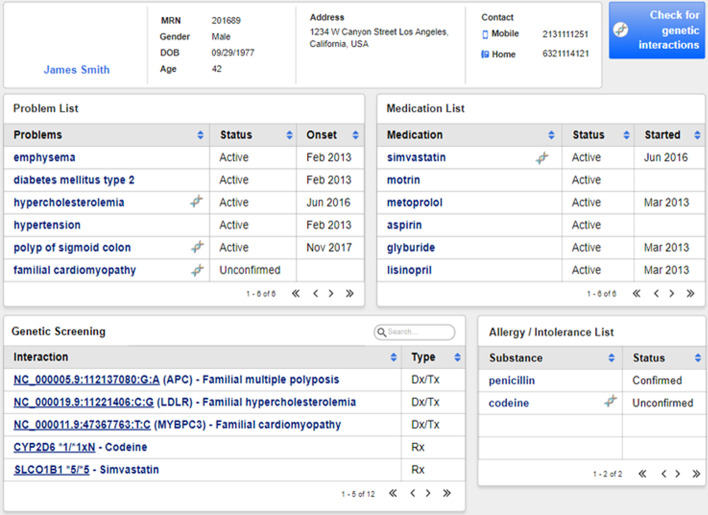
Results: vcf2fhir converts VCF variants into a FHIR Genomics Diagnostic Report. Conversion translates each VCF row into a corresponding FHIR-formatted variant in the generated report. In scope are simple variants (SNVs, MNVs, Indels), along with zygosity and phase relationships, for autosomes, sex chromosomes, and mitochondrial DNA. Input parameters include VCF file and genome build ('GRCh37' or 'GRCh38'); and optionally a conversion region that indicates the region(s) to convert, a studied region that lists genomic regions studied by the lab, and a non-callable region that lists studied regions deemed uncallable by the lab. Conversion can be limited to a subset of VCF by supplying genomic coordinates of the conversion region(s). If studied and non-callable regions are also supplied, the output FHIR report will include 'region-studied' observations that detail which portions of the conversion region were studied, and of those studied regions, which portions were deemed uncallable. We illustrate the vcf2fhir utility via two case studies. The first, 'SMART Cancer Navigator', is a web application that offers clinical decision support by linking patient EHR information to cancerous gene variants. The second, 'Precision Genomics Integration Platform', intersects a patient's FHIR-formatted clinical and genomic data with knowledge bases in order to provide on-demand delivery of contextually relevant genomic findings and recommendations to the EHR.
Conclusions: Experience to date shows that the vcf2fhir utility can be effectively woven into clinically useful genomic-EHR integration pipelines. Additional testing will be a critical step towards the clinical validation of this utility, enabling it to be integrated in a variety of real world data flow scenarios. For now, we propose the use of this utility primarily to accelerate FHIR Genomics understanding and to facilitate experimentation with further integration of genomics data into the EHR.
Keywords: Clinical genomics; EHR integration; FHIR; Next-generation sequencing; SMART-on-FHIR.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- The Variant Call Format Specification. https://samtools.github.io/hts-specs/VCFv4.3.pdf. Accessed 1 May 2020.
-
- HL7 FHIR v4.0.1. https://www.hl7.org/fhir/. Accessed 15 Oct 2020.
-
- Phenopackets: Standardizing and Exchanging Patient Phenotypic Data. https://www.ga4gh.org/news/phenopackets-standardizing-and-exchanging-pat.... Accessed 15 Oct 2020.
-
- HL7 FHIR Genomics Reporting Implementation Guide. http://hl7.org/fhir/uv/genomics-reporting/index.html. Accessed 15 Oct 2020.
-
- HL7 FHIR mCode Implementation Guide. http://hl7.org/fhir/us/mcode/. Accessed 15 Oct 2020.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
