

Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology
- PMID: 33654267
- PMCID: PMC7925608
- DOI: 10.1038/s41698-021-00155-6
Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology
Abstract
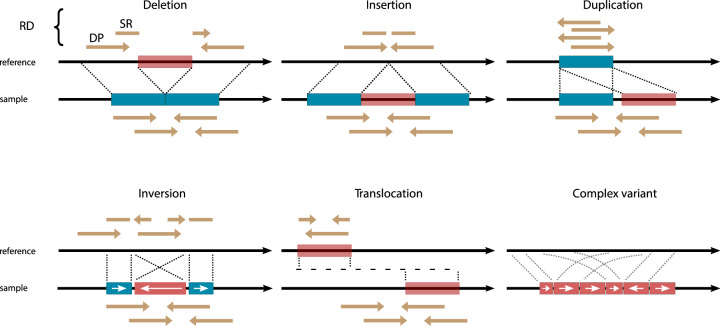
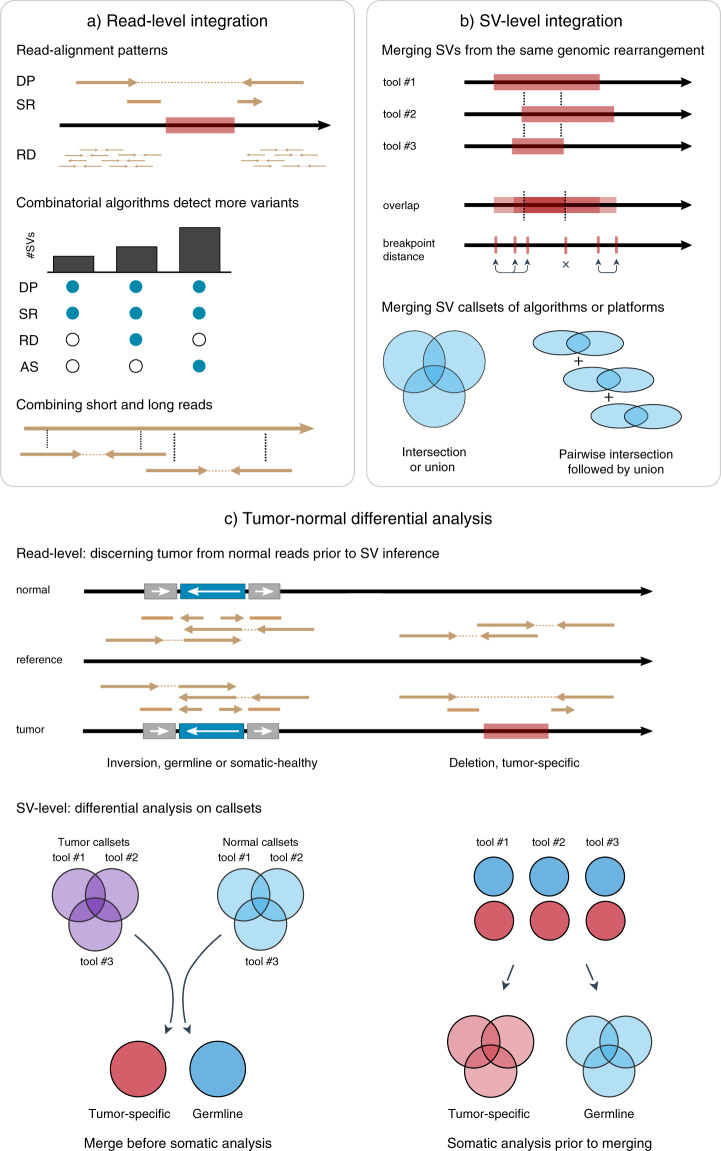
Cancer is generally characterized by acquired genomic aberrations in a broad spectrum of types and sizes, ranging from single nucleotide variants to structural variants (SVs). At least 30% of cancers have a known pathogenic SV used in diagnosis or treatment stratification. However, research into the role of SVs in cancer has been limited due to difficulties in detection. Biological and computational challenges confound SV detection in cancer samples, including intratumor heterogeneity, polyploidy, and distinguishing tumor-specific SVs from germline and somatic variants present in healthy cells. Classification of tumor-specific SVs is challenging due to inconsistencies in detected breakpoints, derived variant types and biological complexity of some rearrangements. Full-spectrum SV detection with high recall and precision requires integration of multiple algorithms and sequencing technologies to rescue variants that are difficult to resolve through individual methods. Here, we explore current strategies for integrating SV callsets and to enable the use of tumor-specific SVs in precision oncology.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
