

Yeast Single-cell RNA-seq, Cell by Cell and Step by Step
- PMID: 33654857
- PMCID: PMC7854150
- DOI: 10.21769/BioProtoc.3359
Yeast Single-cell RNA-seq, Cell by Cell and Step by Step
Abstract
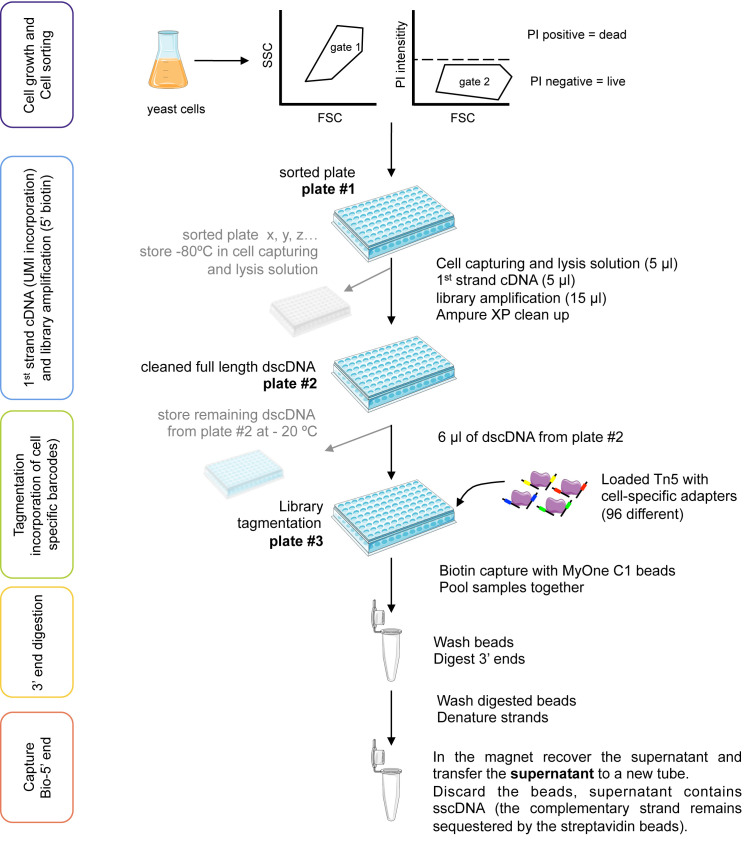
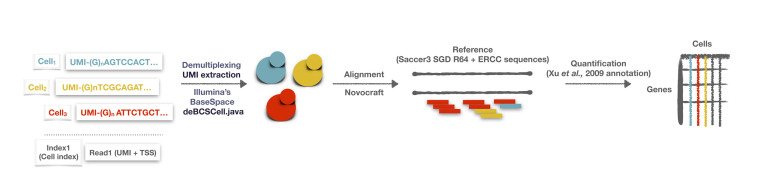
Single-cell RNA-seq (scRNA-seq) has become an established method for uncovering the intrinsic complexity within populations. Even within seemingly homogenous populations of isogenic yeast cells, there is a high degree of heterogeneity that originates from a compact and pervasively transcribed genome. Research with microorganisms such as yeast represents a major challenge for single-cell transcriptomics, due to their small size, rigid cell wall, and low RNA content per cell. Because of these technical challenges, yeast-specific scRNA-seq methodologies have recently started to appear, each one of them relying on different cell-isolation and library-preparation methods. Consequently, each approach harbors unique strengths and weaknesses that need to be considered. We have recently developed a yeast single-cell RNA-seq protocol (yscRNA-seq), which is inexpensive, high-throughput and easy-to-implement, tailored to the unique needs of yeast. yscRNA-seq provides a unique platform that combines single-cell phenotyping via index sorting with the incorporation of unique molecule identifiers on transcripts that allows to digitally count the number of molecules in a strand- and isoform-specific manner. Here, we provide a detailed, step-by-step description of the experimental and computational steps of yscRNA-seq protocol. This protocol will ease the implementation of yscRNA-seq in other laboratories and provide guidelines for the development of novel technologies.
Keywords: Noncoding RNA; Single-cell RNA-seq; Transcript isoforms; Transcriptomics; Yeast.
Copyright © 2019 The Authors; exclusive licensee Bio-protocol LLC.
Conflict of interest statement
Competing interestsThe authors declare no financial or non-financial interests.
Figures
References
-
- Gasch A. P., Yu F. B., Hose J., Escalante L. E., Place M., Bacher R., Kanbar J., Ciobanu D., Sandor L., Grigoriev I. V., Kendziorski C., Quake S. R. and McClean M. N.(2017). Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol 15(12): e2004050. - PMC - PubMed
