

Validation strategy of a bioinformatics whole genome sequencing workflow for Shiga toxin-producing Escherichia coli using a reference collection extensively characterized with conventional methods
- PMID: 33656437
- PMCID: PMC8190621
- DOI: 10.1099/mgen.0.000531
Validation strategy of a bioinformatics whole genome sequencing workflow for Shiga toxin-producing Escherichia coli using a reference collection extensively characterized with conventional methods
Abstract
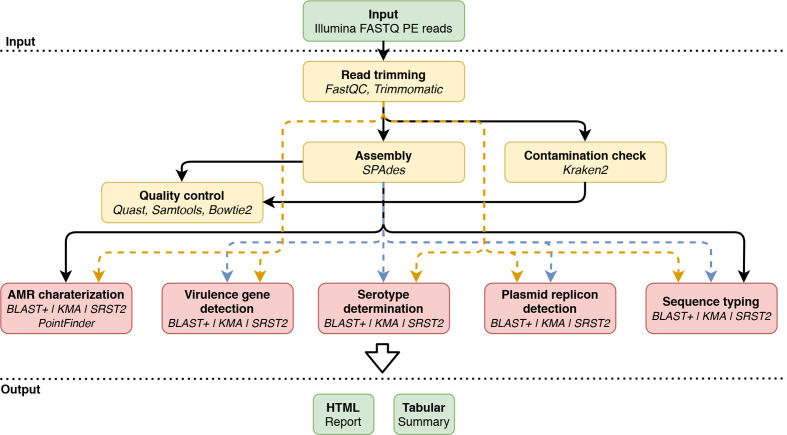
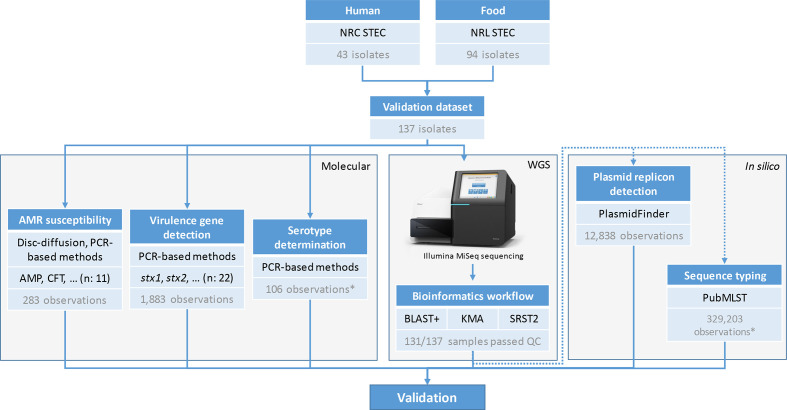
Whole genome sequencing (WGS) enables complete characterization of bacterial pathogenic isolates at single nucleotide resolution, making it the ultimate tool for routine surveillance and outbreak investigation. The lack of standardization, and the variation regarding bioinformatics workflows and parameters, however, complicates interoperability among (inter)national laboratories. We present a validation strategy applied to a bioinformatics workflow for Illumina data that performs complete characterization of Shiga toxin-producing Escherichia coli (STEC) isolates including antimicrobial resistance prediction, virulence gene detection, serotype prediction, plasmid replicon detection and sequence typing. The workflow supports three commonly used bioinformatics approaches for the detection of genes and alleles: alignment with blast+, kmer-based read mapping with KMA, and direct read mapping with SRST2. A collection of 131 STEC isolates collected from food and human sources, extensively characterized with conventional molecular methods, was used as a validation dataset. Using a validation strategy specifically adopted to WGS, we demonstrated high performance with repeatability, reproducibility, accuracy, precision, sensitivity and specificity above 95 % for the majority of all assays. The WGS workflow is publicly available as a 'push-button' pipeline at https://galaxy.sciensano.be. Our validation strategy and accompanying reference dataset consisting of both conventional and WGS data can be used for characterizing the performance of various bioinformatics workflows and assays, facilitating interoperability between laboratories with different WGS and bioinformatics set-ups.
Keywords: Escherichia coli; STEC, foodborne pathogens; public health; validation; whole genome sequencing.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

References
-
- Carriço JA, Sabat AJ, Friedrich AW, Ramirez M. Bioinformatics in bacterial molecular epidemiology and public health: databases, tools and the next-generation sequencing revolution, on behalf of the ESCMID Study Group for Epidemiological Markers (ESGEM) Eurosurveillance. 2013;18:1–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials