

Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1
- PMID: 33656802
- PMCID: PMC8235124
- DOI: 10.1002/sctm.20-0386
Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1
Abstract
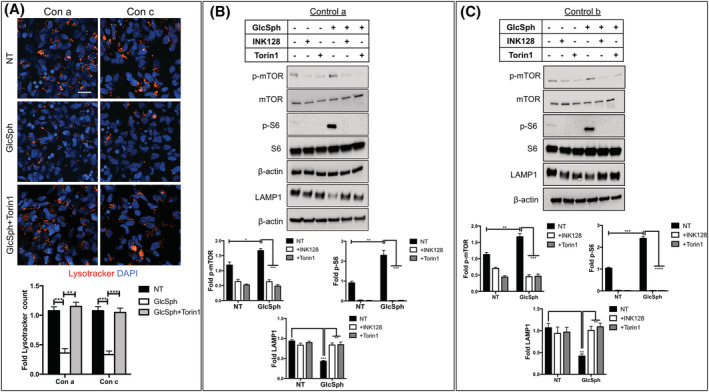
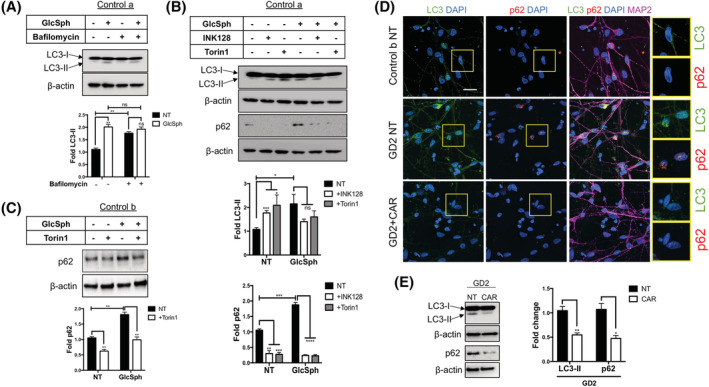
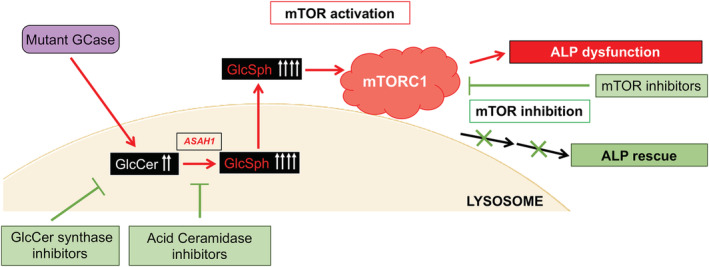
Gaucher disease (GD) is a lysosomal storage disorder caused by mutations in GBA1, the gene that encodes lysosomal β-glucocerebrosidase (GCase). Mild mutations in GBA1 cause type 1 non-neuronopathic GD, whereas severe mutations cause types 2 and 3 neuronopathic GD (nGD). GCase deficiency results in the accumulation of glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph). GlcSph is formed by deacylation of GlcCer by the lysosomal enzyme acid ceramidase. Brains from patients with nGD have high levels of GlcSph, a lipid believed to play an important role in nGD, but the mechanisms involved remain unclear. To identify these mechanisms, we used human induced pluripotent stem cell-derived neurons from nGD patients. We found that elevated levels of GlcSph activate mammalian target of rapamycin (mTOR) complex 1 (mTORC1), interfering with lysosomal biogenesis and autophagy, which were restored by incubation of nGD neurons with mTOR inhibitors. We also found that inhibition of acid ceramidase prevented both, mTOR hyperactivity and lysosomal dysfunction, suggesting that these alterations were caused by GlcSph accumulation in the mutant neurons. To directly determine whether GlcSph can cause mTOR hyperactivation, we incubated wild-type neurons with exogenous GlcSph. Remarkably, GlcSph treatment recapitulated the mTOR hyperactivation and lysosomal abnormalities in mutant neurons, which were prevented by coincubation of GlcSph with mTOR inhibitors. We conclude that elevated GlcSph activates an mTORC1-dependent pathogenic mechanism that is responsible for the lysosomal abnormalities of nGD neurons. We also identify acid ceramidase as essential to the pathogenesis of nGD, providing a new therapeutic target for treating GBA1-associated neurodegeneration.
Keywords: drug target; experimental models; induced pluripotent stem cells; neural differentiation; neuropathy; signal transduction; stem/progenitor cell.
© 2021 The Authors. STEM CELLS TRANSLATIONAL MEDICINE published by Wiley Periodicals LLC on behalf of AlphaMed Press.
Conflict of interest statement
The authors declared no potential conflicts of interest.
Figures
Similar articles
-
Acid ceramidase involved in pathogenic cascade leading to accumulation of α-synuclein in iPSC model of GBA1-associated Parkinson's disease.Hum Mol Genet. 2023 May 18;32(11):1888-1900. doi: 10.1093/hmg/ddad025. Hum Mol Genet. 2023. PMID: 36752535 Free PMC article.
-
Novel beta-glucocerebrosidase chaperone compounds identified from cell-based screening reduce pathologically accumulated glucosylsphingosine in iPS-derived neuronal cells.SLAS Discov. 2023 Oct;28(7):344-349. doi: 10.1016/j.slasd.2023.06.002. Epub 2023 Jun 25. SLAS Discov. 2023. PMID: 37369311
-
mTOR hyperactivity mediates lysosomal dysfunction in Gaucher's disease iPSC-neuronal cells.Dis Model Mech. 2019 Oct 16;12(10):dmm038596. doi: 10.1242/dmm.038596. Dis Model Mech. 2019. PMID: 31519738 Free PMC article.
-
Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease.Adv Drug Deliv Rev. 2022 Aug;187:114402. doi: 10.1016/j.addr.2022.114402. Epub 2022 Jun 25. Adv Drug Deliv Rev. 2022. PMID: 35764179 Review.
-
Animal Models for the Study of Gaucher Disease.Int J Mol Sci. 2023 Nov 7;24(22):16035. doi: 10.3390/ijms242216035. Int J Mol Sci. 2023. PMID: 38003227 Free PMC article. Review.
Cited by
-
Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases.Biomedicines. 2023 Apr 1;11(4):1067. doi: 10.3390/biomedicines11041067. Biomedicines. 2023. PMID: 37189685 Free PMC article. Review.
-
A review on Gaucher disease: therapeutic potential of β-glucocerebrosidase-targeted mRNA/saRNA approach.Int J Biol Sci. 2024 Mar 17;20(6):2111-2129. doi: 10.7150/ijbs.87741. eCollection 2024. Int J Biol Sci. 2024. PMID: 38617529 Free PMC article. Review.
-
Deficiency in NPC2 results in disruption of mitochondria-late endosome/lysosomes contact sites and endo-lysosomal lipid dyshomeostasis.Sci Rep. 2025 Jan 2;15(1):325. doi: 10.1038/s41598-024-83460-x. Sci Rep. 2025. PMID: 39747180 Free PMC article.
-
Glucocerebrosidase mutations and Parkinson disease.J Neural Transm (Vienna). 2022 Sep;129(9):1105-1117. doi: 10.1007/s00702-022-02531-3. Epub 2022 Aug 6. J Neural Transm (Vienna). 2022. PMID: 35932311 Free PMC article. Review.
-
C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages.Int J Mol Sci. 2021 Sep 14;22(18):9912. doi: 10.3390/ijms22189912. Int J Mol Sci. 2021. PMID: 34576075 Free PMC article.
References
-
- Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J Neurochem. 1982;39(3):709‐718. - PubMed
-
- Orvisky E, Sidransky E, McKinney CE, et al. Glucosylsphingosine accumulation in mice and patients with type 2 Gaucher disease begins early in gestation. Pediatr Res. 2000;48(2):233‐237. - PubMed
-
- Sidransky E. Gaucher disease: complexity in a "simple" disorder. Mol Genet Metab. 2004;83(1‐2):6‐15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
