

COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas
- PMID: 33657410
- PMCID: PMC7857060
- DOI: 10.1016/j.cell.2021.01.053
COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas
Erratum in
-
COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas.Cell. 2021 Nov 11;184(23):5838. doi: 10.1016/j.cell.2021.10.023. Cell. 2021. PMID: 34767776 Free PMC article. No abstract available.
Abstract
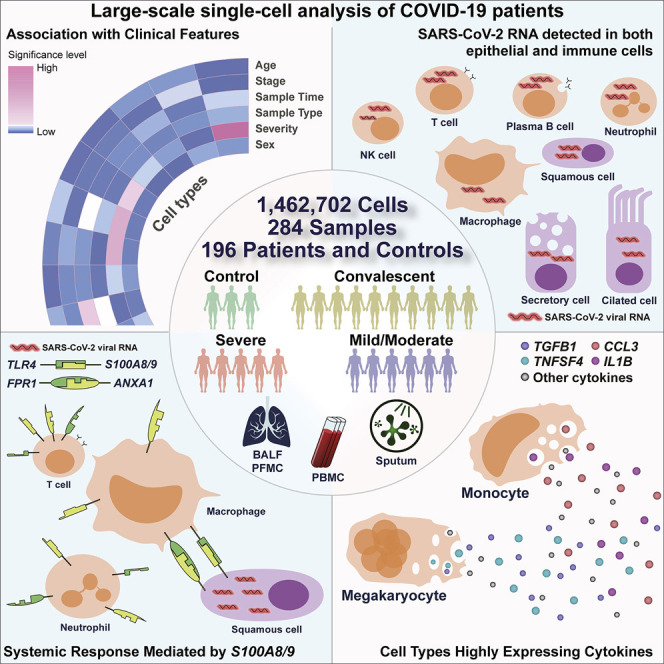
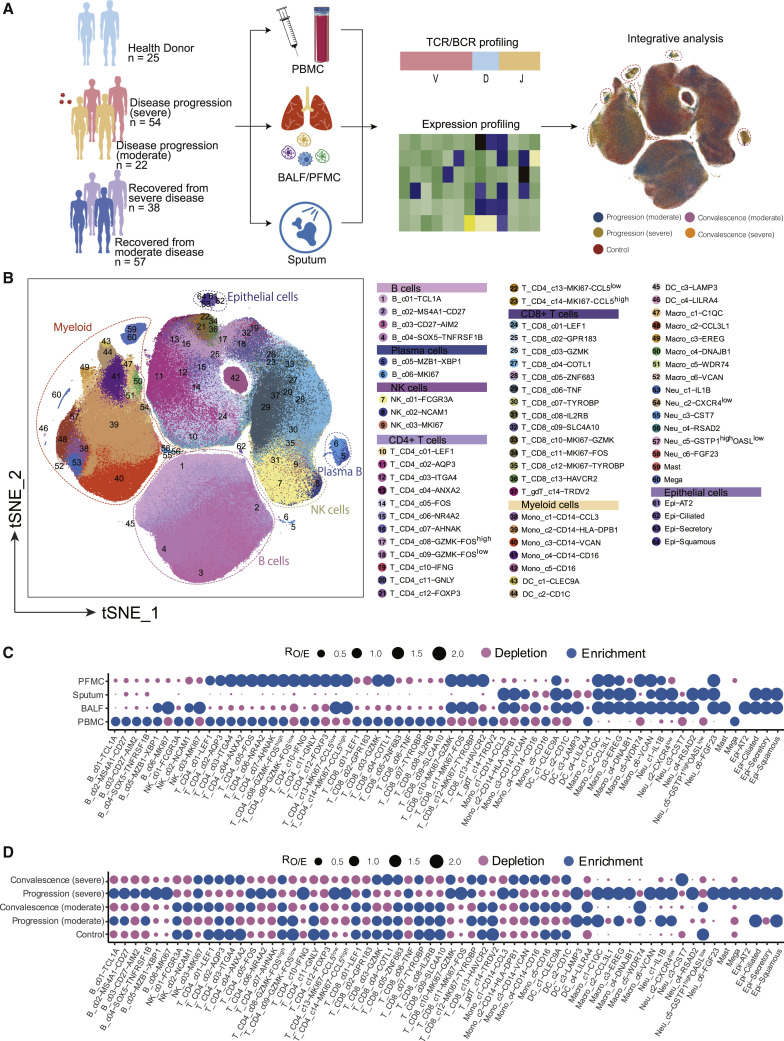
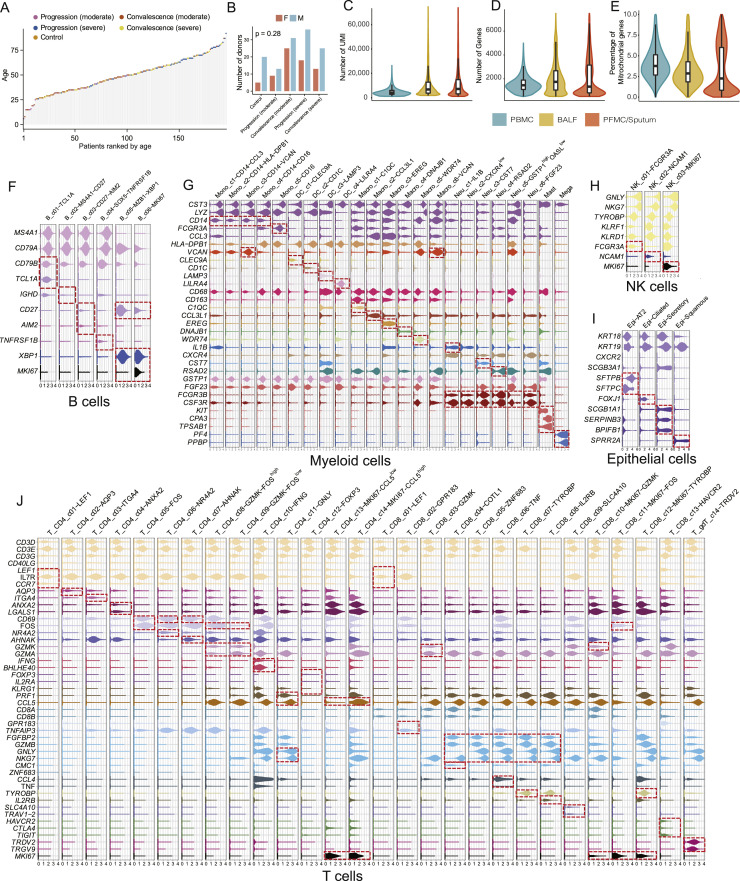
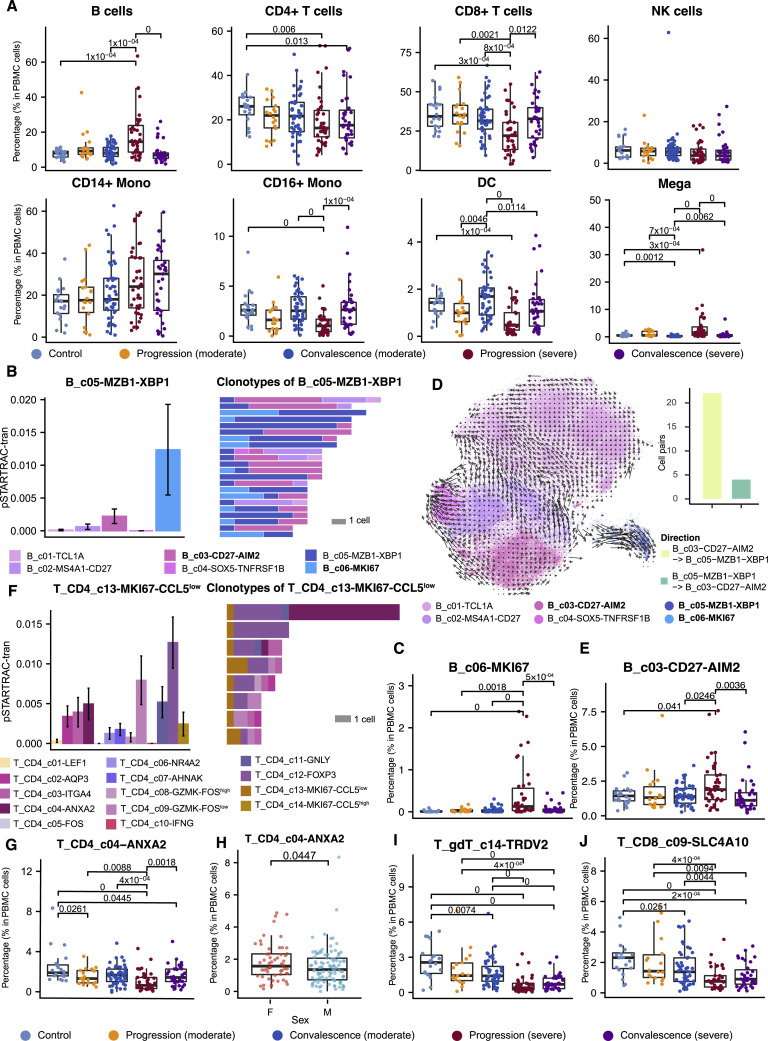
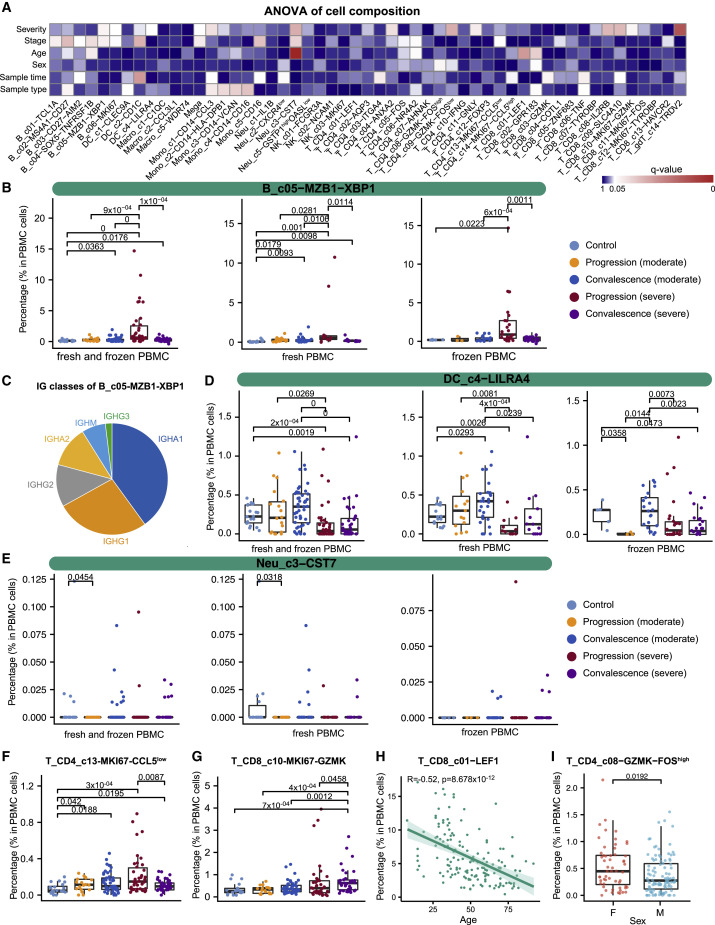
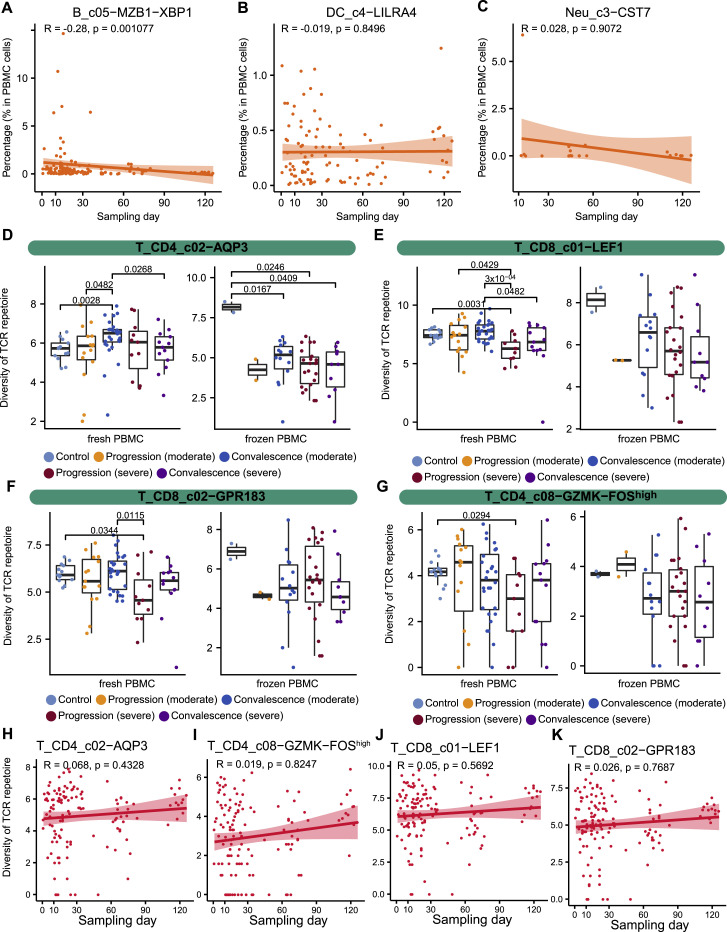
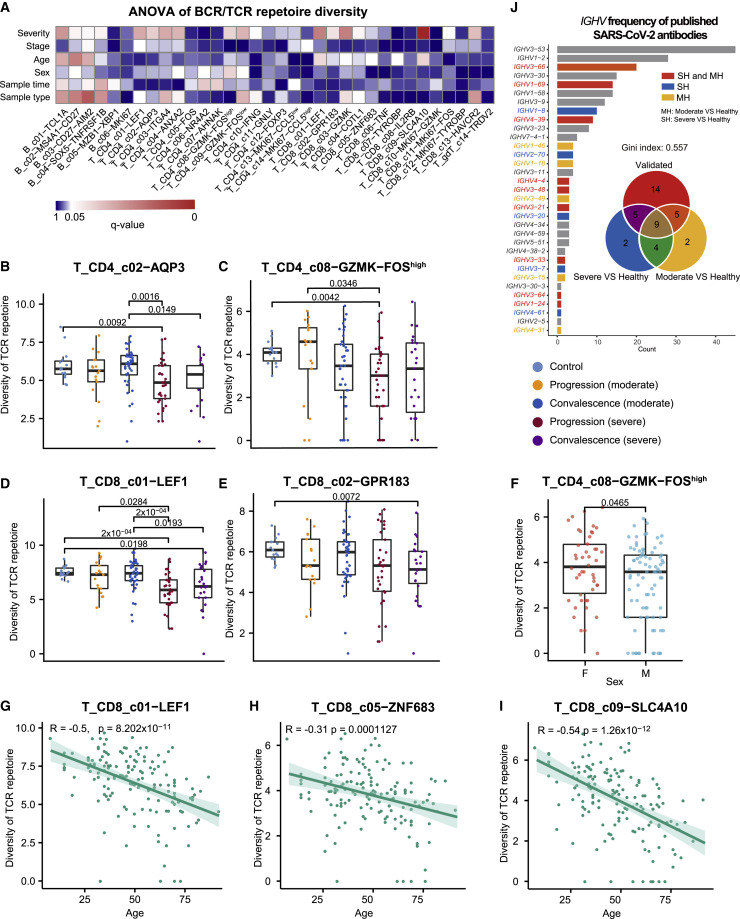
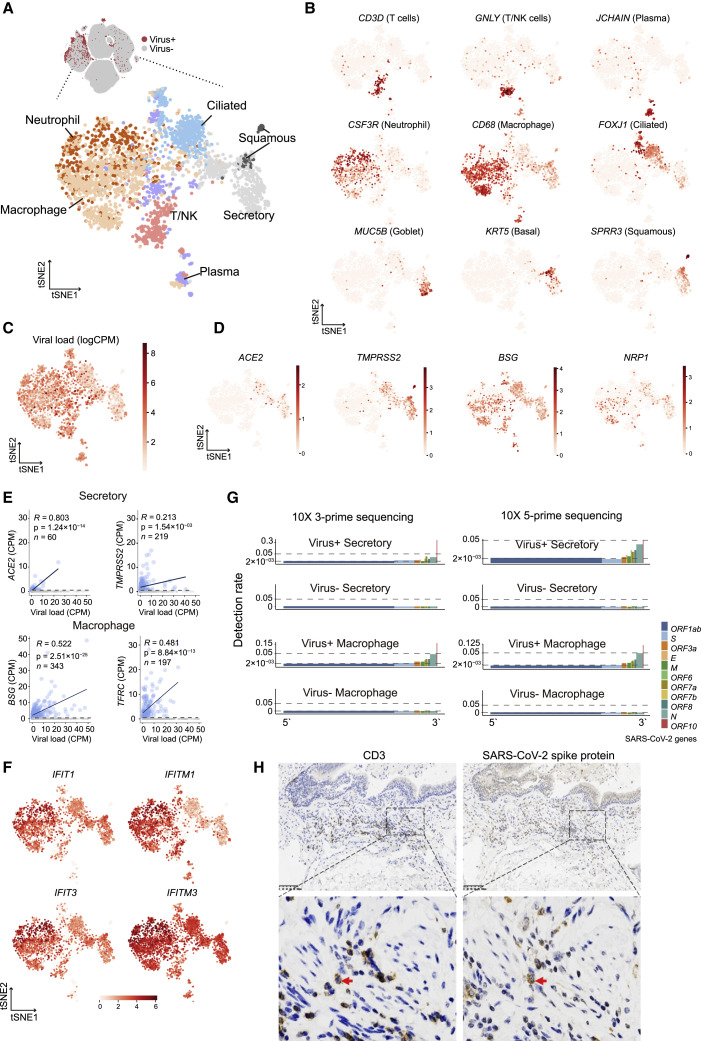
A dysfunctional immune response in coronavirus disease 2019 (COVID-19) patients is a recurrent theme impacting symptoms and mortality, yet a detailed understanding of pertinent immune cells is not complete. We applied single-cell RNA sequencing to 284 samples from 196 COVID-19 patients and controls and created a comprehensive immune landscape with 1.46 million cells. The large dataset enabled us to identify that different peripheral immune subtype changes are associated with distinct clinical features, including age, sex, severity, and disease stages of COVID-19. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA was found in diverse epithelial and immune cell types, accompanied by dramatic transcriptomic changes within virus-positive cells. Systemic upregulation of S100A8/A9, mainly by megakaryocytes and monocytes in the peripheral blood, may contribute to the cytokine storms frequently observed in severe patients. Our data provide a rich resource for understanding the pathogenesis of and developing effective therapeutic strategies for COVID-19.
Keywords: B cell receptor sequencing; COVID-19; SARS-CoV-2; T cell receptor sequencing; cell-cell interaction; cytokine storm; host cell range; ligand-receptor interaction; single-cell RNA-seq; single-cell transcriptomics.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Zemin Zhang is a founder of Analytical Bioscience and an advisor for InnoCare. All financial interests are unrelated to this study. The remining authors declare no competing interests.
Figures

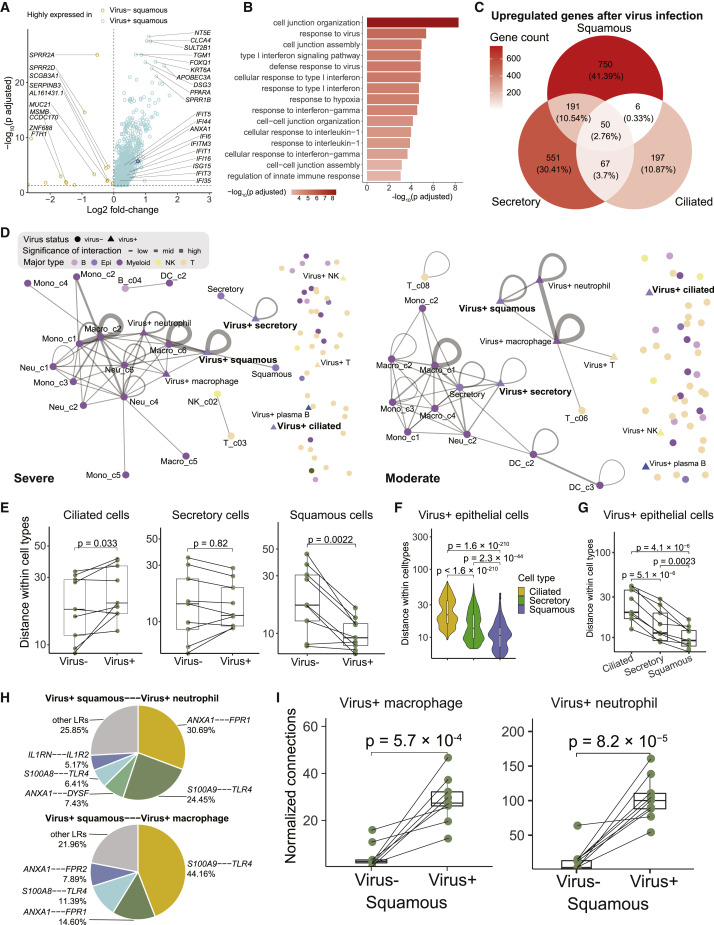
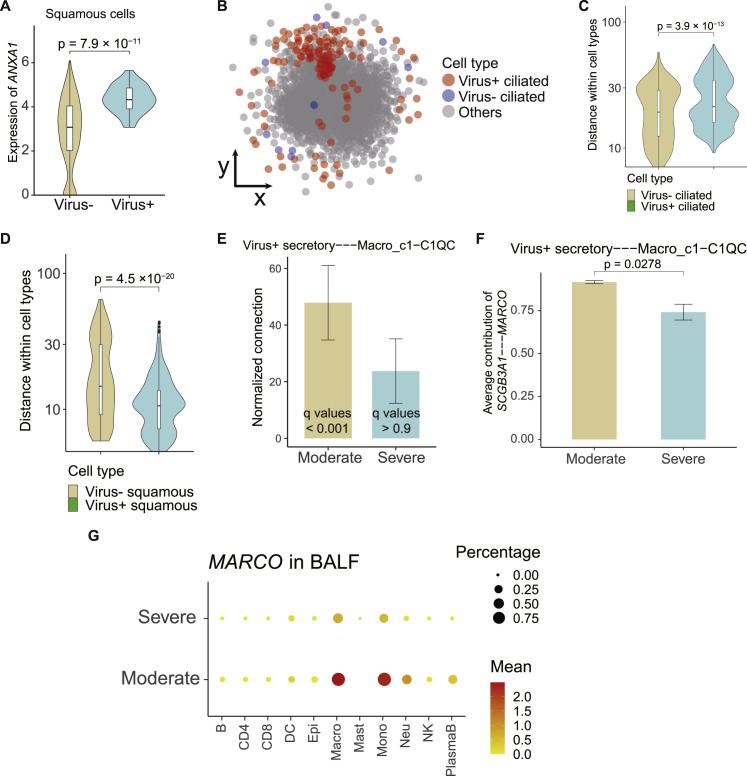

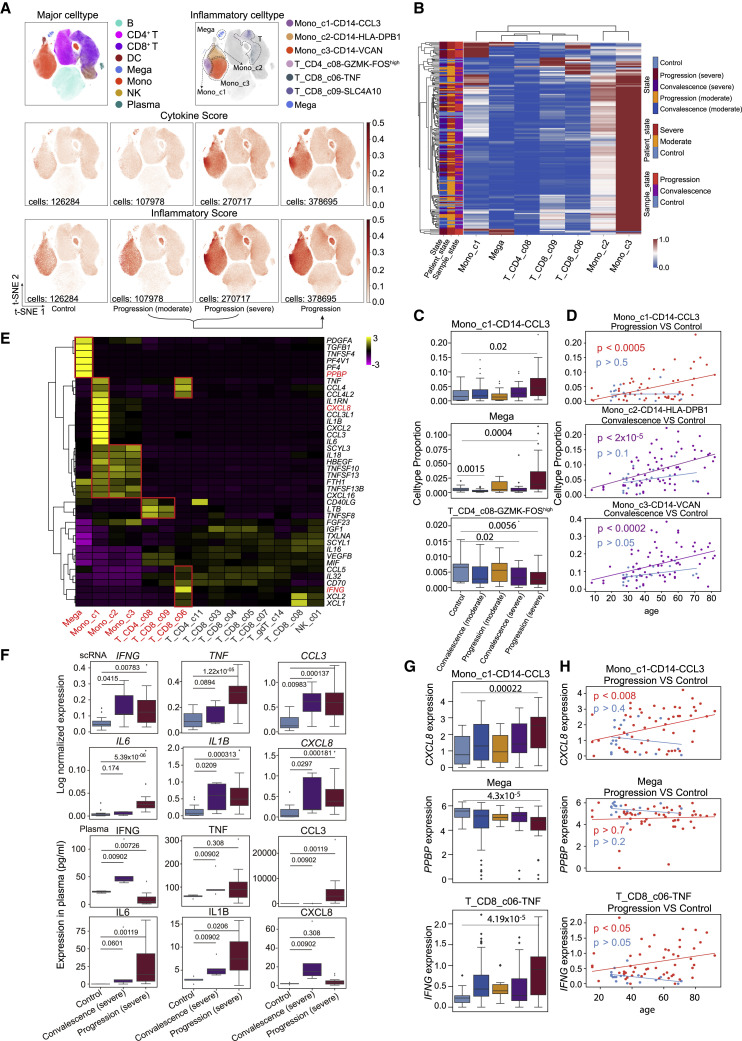
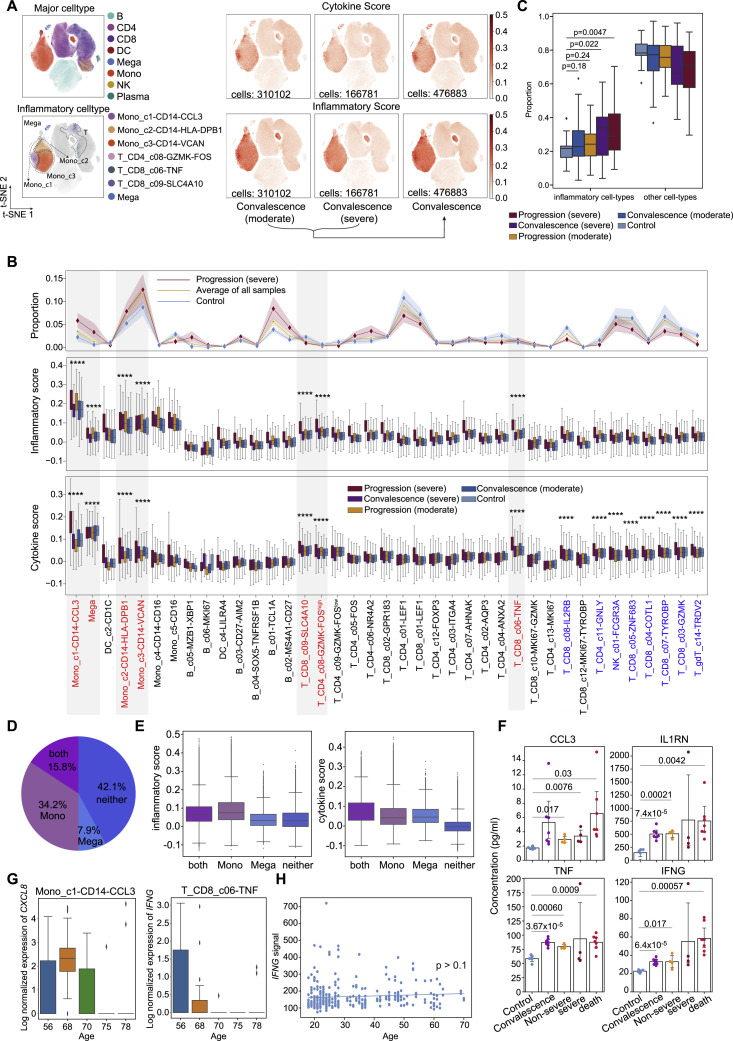
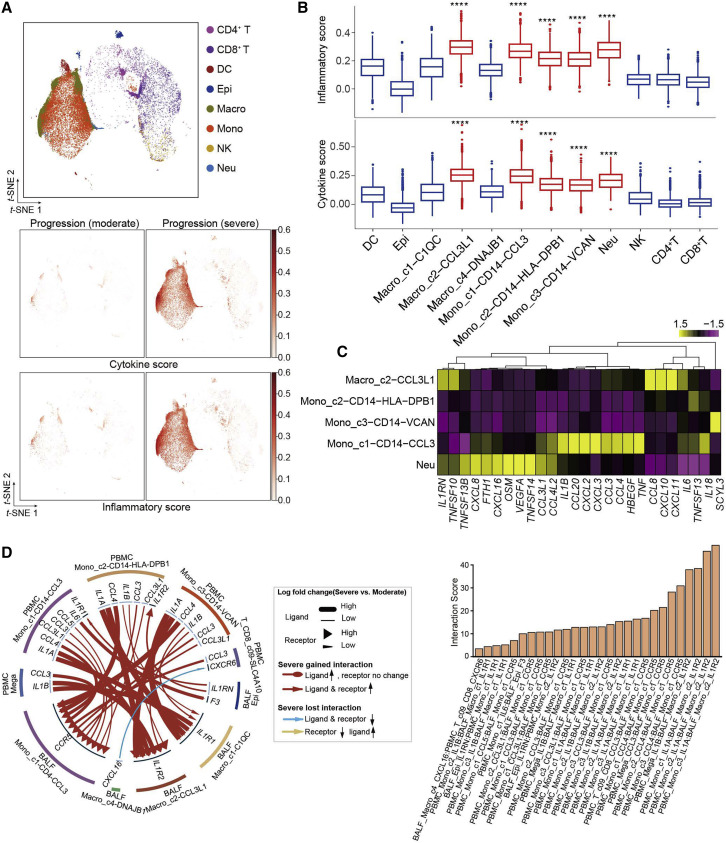
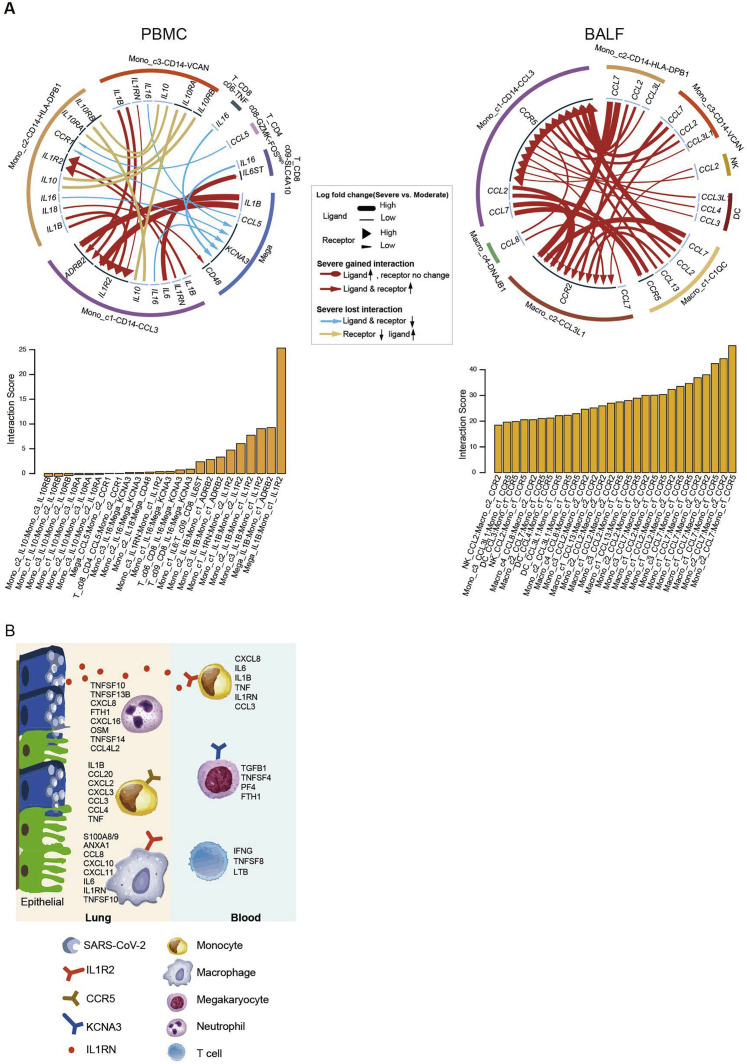
Comment in
-
COVID-19: complexity of disease severity revealed by systemic and localized single cell immune atlas.Signal Transduct Target Ther. 2021 Apr 16;6(1):156. doi: 10.1038/s41392-021-00587-3. Signal Transduct Target Ther. 2021. PMID: 33863870 Free PMC article. No abstract available.
References
-
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
