

SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration
- PMID: 33657413
- PMCID: PMC8174188
- DOI: 10.1016/j.neuron.2021.02.009
SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration
Abstract
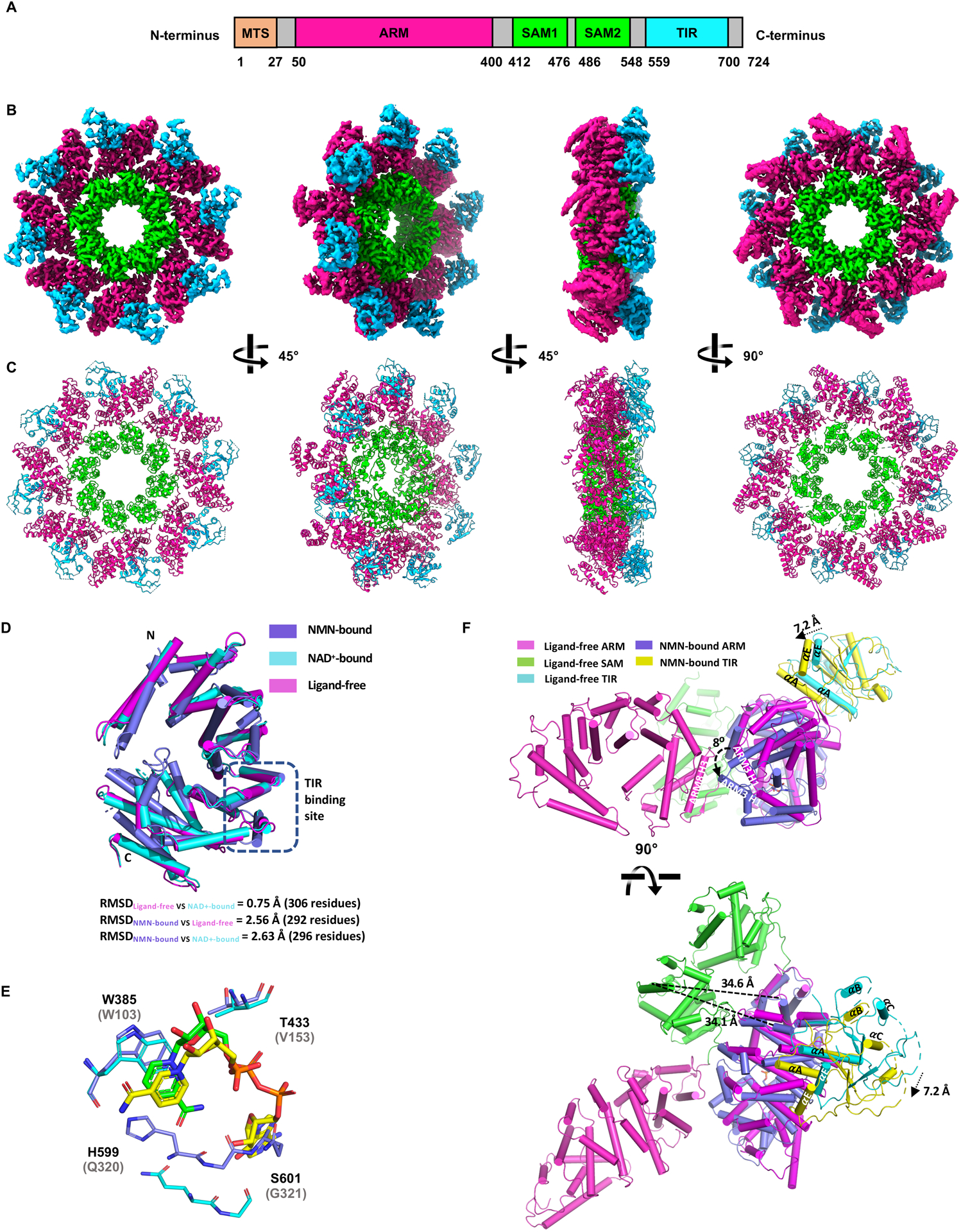
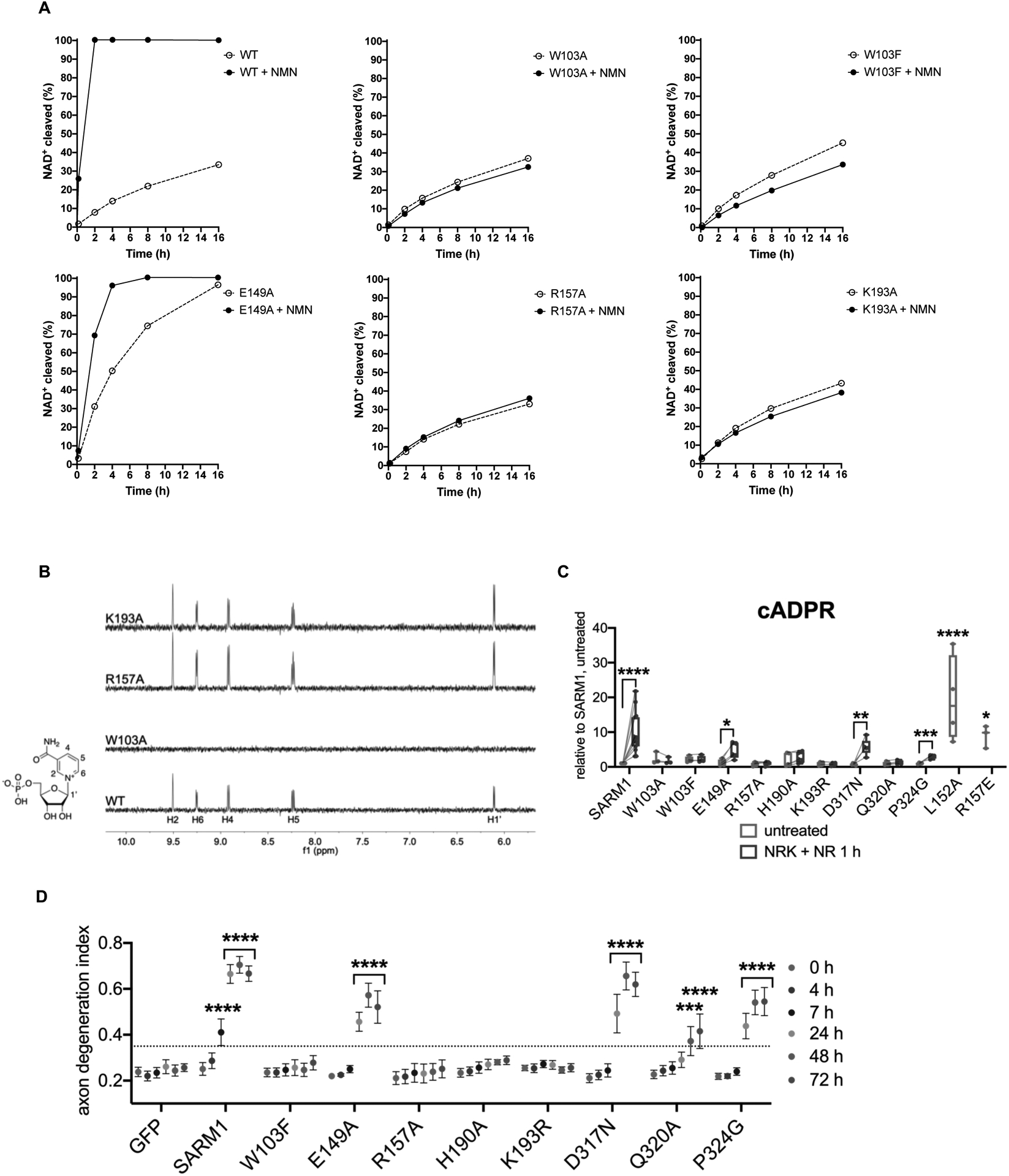
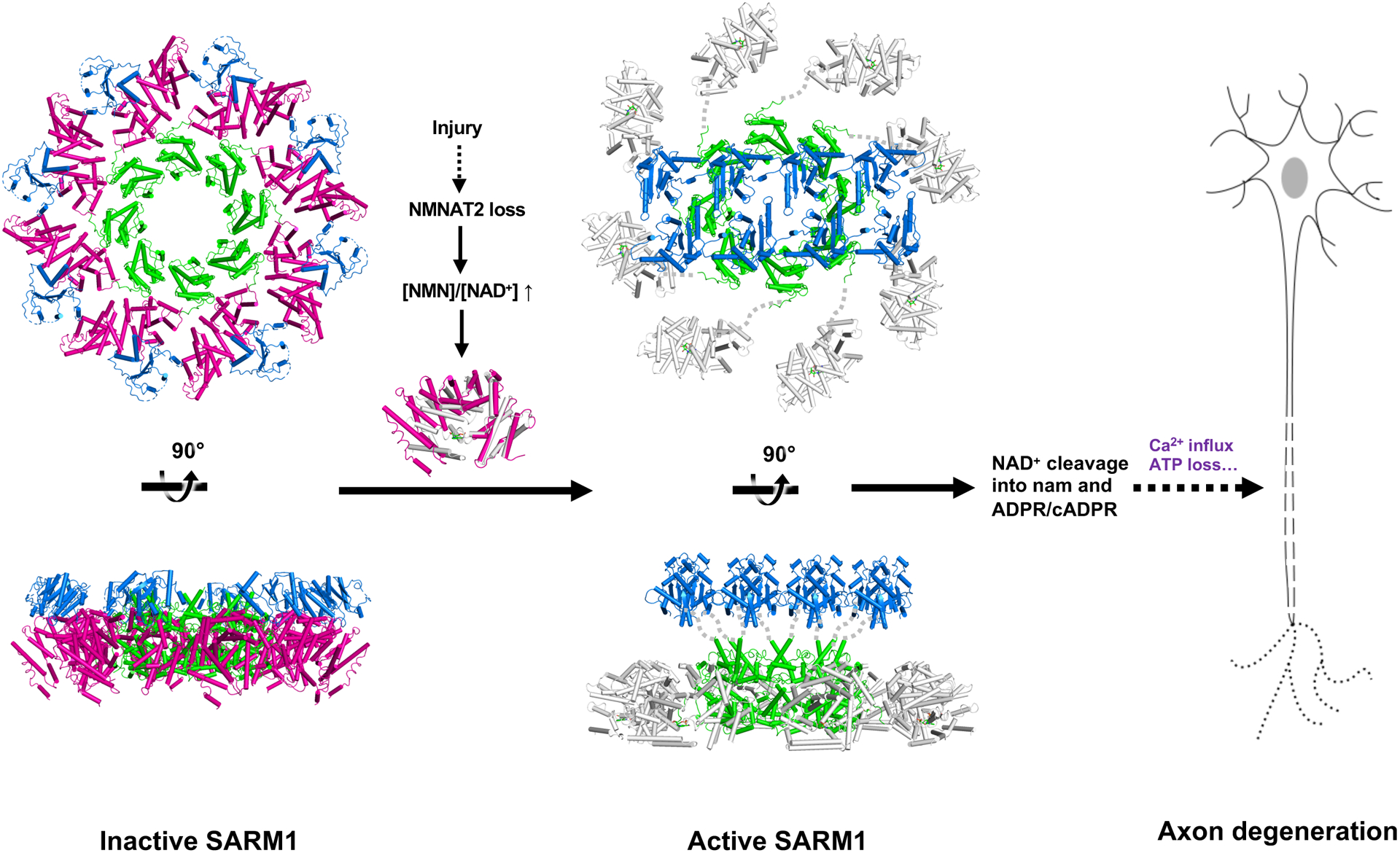
Axon degeneration is a central pathological feature of many neurodegenerative diseases. Sterile alpha and Toll/interleukin-1 receptor motif-containing 1 (SARM1) is a nicotinamide adenine dinucleotide (NAD+)-cleaving enzyme whose activation triggers axon destruction. Loss of the biosynthetic enzyme NMNAT2, which converts nicotinamide mononucleotide (NMN) to NAD+, activates SARM1 via an unknown mechanism. Using structural, biochemical, biophysical, and cellular assays, we demonstrate that SARM1 is activated by an increase in the ratio of NMN to NAD+ and show that both metabolites compete for binding to the auto-inhibitory N-terminal armadillo repeat (ARM) domain of SARM1. We report structures of the SARM1 ARM domain bound to NMN and of the homo-octameric SARM1 complex in the absence of ligands. We show that NMN influences the structure of SARM1 and demonstrate via mutagenesis that NMN binding is required for injury-induced SARM1 activation and axon destruction. Hence, SARM1 is a metabolic sensor responding to an increased NMN/NAD+ ratio by cleaving residual NAD+, thereby inducing feedforward metabolic catastrophe and axonal demise.
Keywords: ARM domain; NADase; TIR domain; X-ray crystallography; allostery; cryo-EM; nicotinamide riboside.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.D. and J.M. are co-founders, scientific advisory board members, and shareholders of Disarm Therapeutics. B.K. is shareholder of Disarm Therapeutics. Y. Sasaki and B.K. are consultants to Disarm Therapeutics. B.K. and T.V. receive research funding from Disarm Therapeutics. R.O.H. and T.B. are employees and shareholders in Disarm Therapeutics. K.C. and P.S.K. are employees of Evotec (UK) Ltd. The authors declare no additional competing interests.
Figures
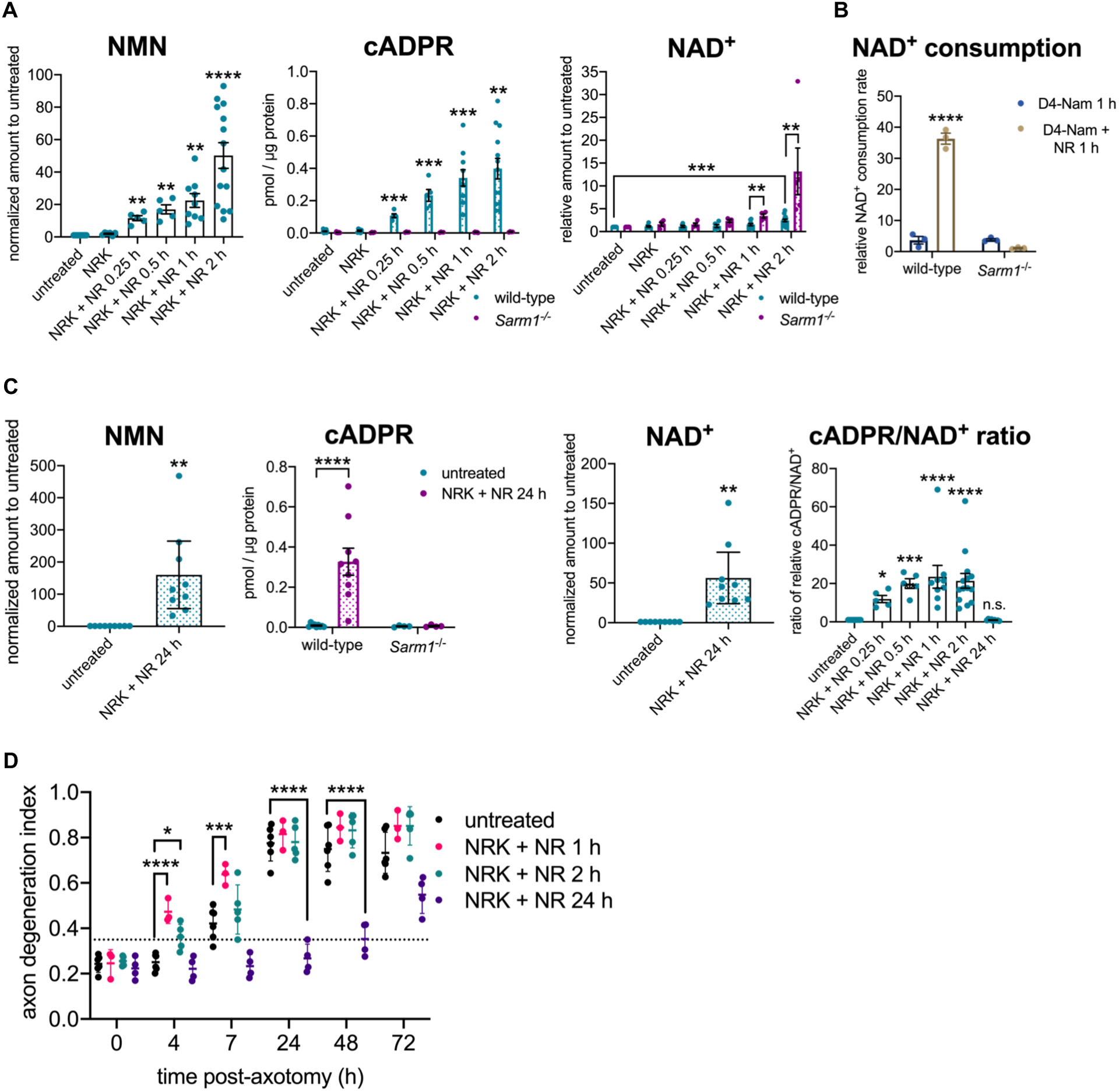
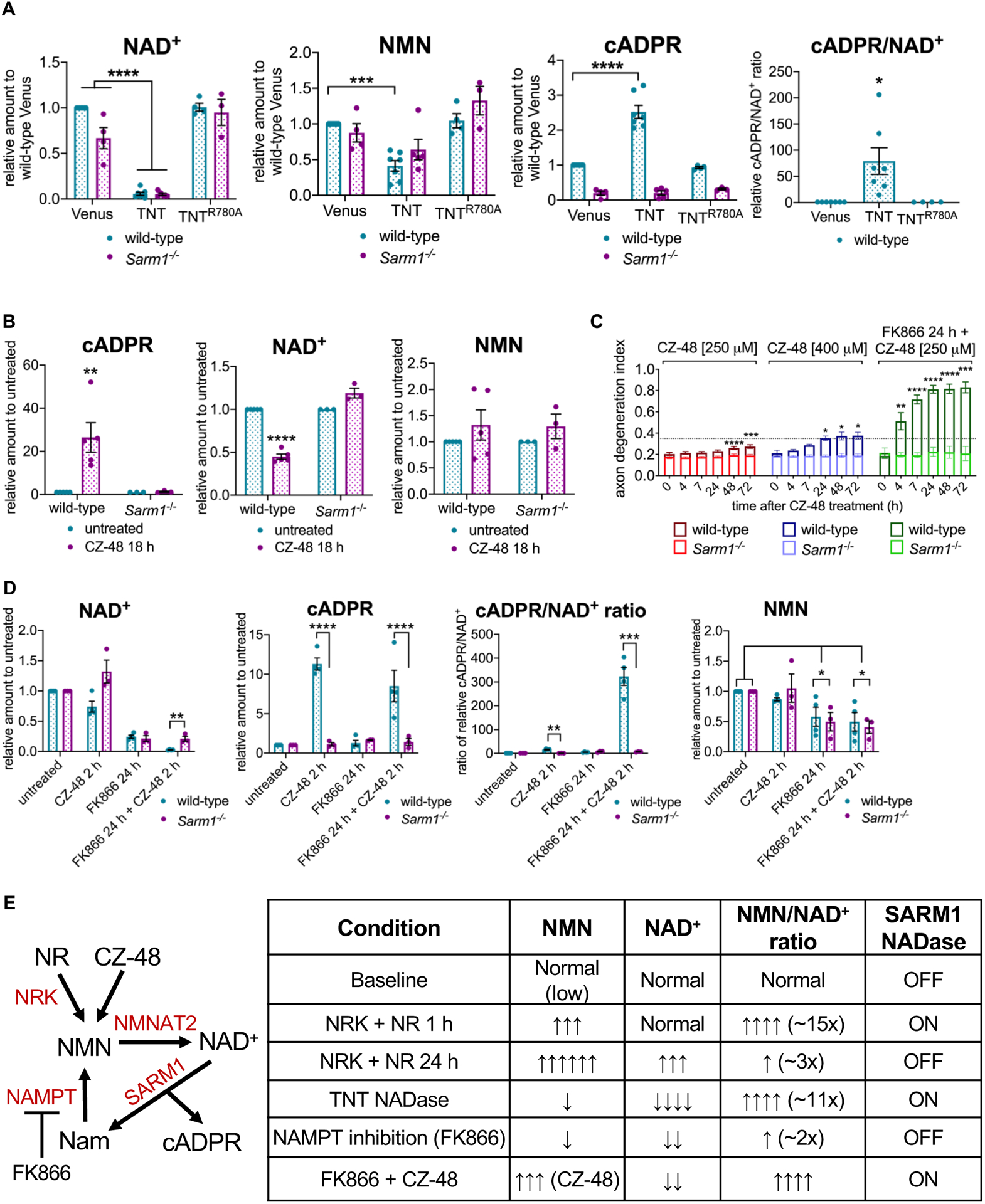
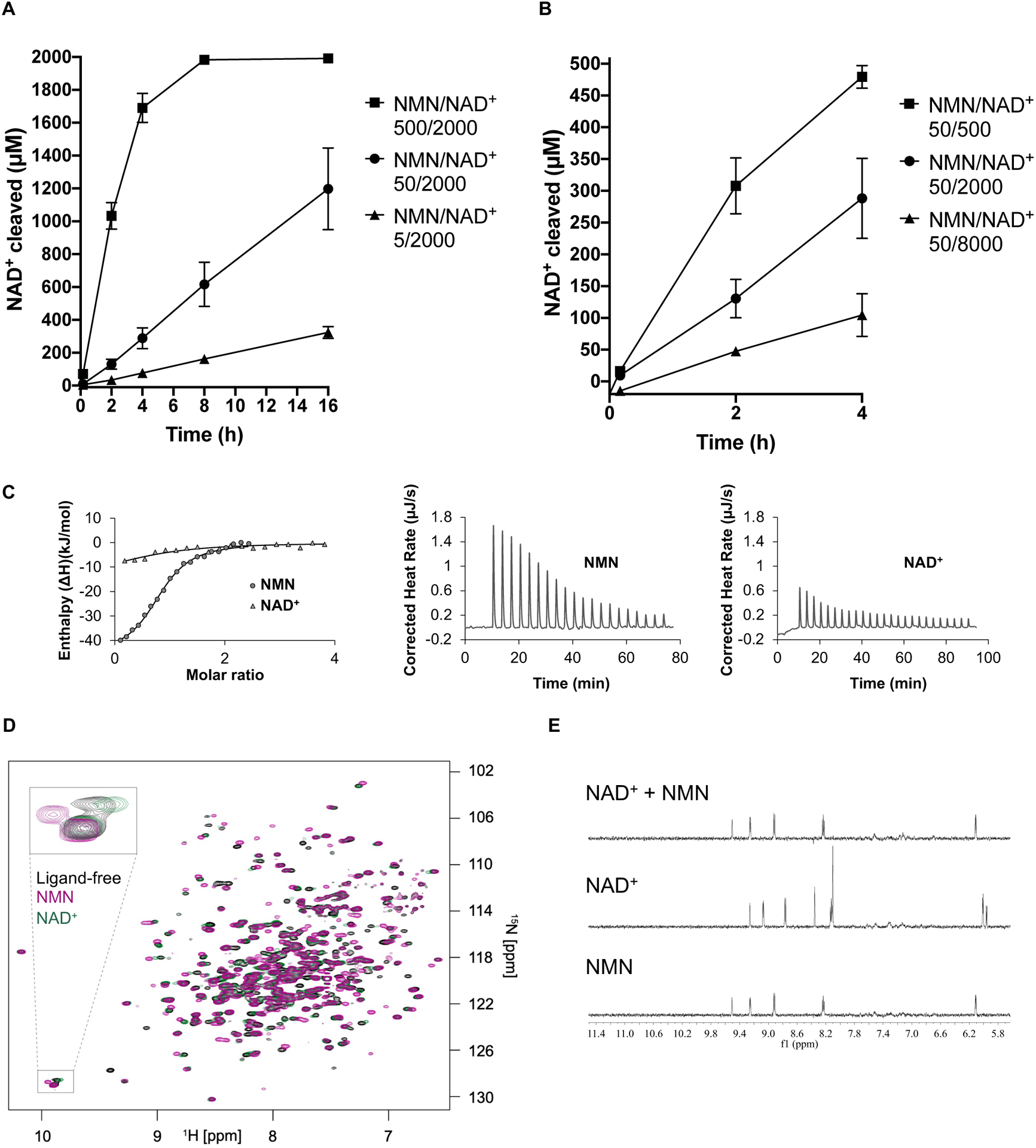
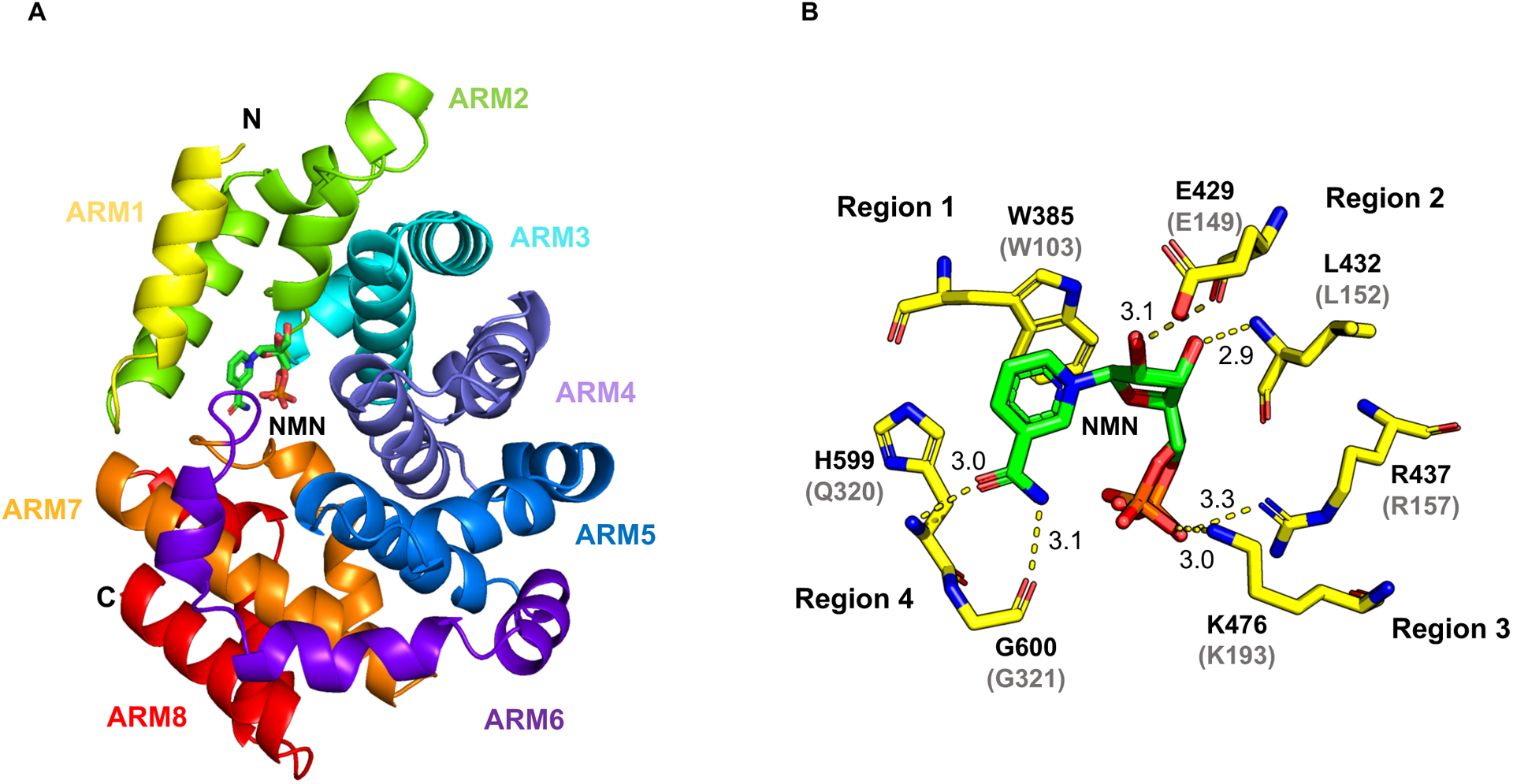
Comment in
-
An NAD+/NMN balancing act by SARM1 and NMNAT2 controls axonal degeneration.Neuron. 2021 Apr 7;109(7):1067-1069. doi: 10.1016/j.neuron.2021.03.021. Neuron. 2021. PMID: 33831359 Free PMC article.
References
-
- Araki T, Sasaki Y, and Milbrandt J (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. - PubMed
-
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys 81, 3684–3690.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
