

Integrative pan cancer analysis reveals epigenomic variation in cancer type and cell specific chromatin domains
- PMID: 33658503
- PMCID: PMC7930052
- DOI: 10.1038/s41467-021-21707-1
Integrative pan cancer analysis reveals epigenomic variation in cancer type and cell specific chromatin domains
Abstract
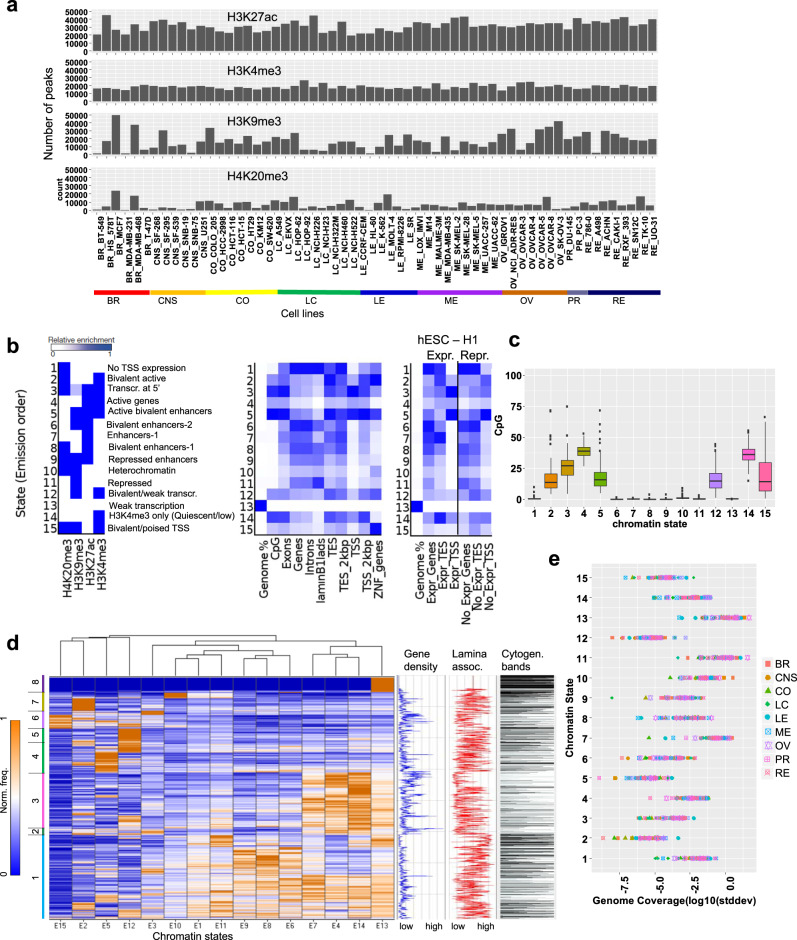
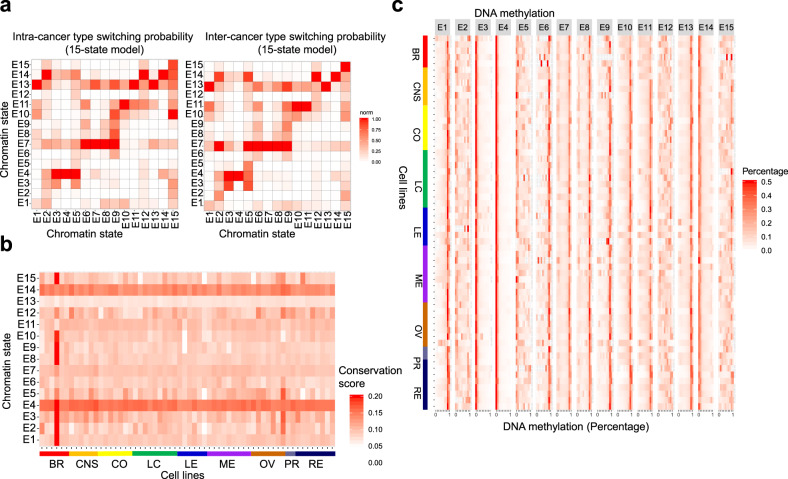
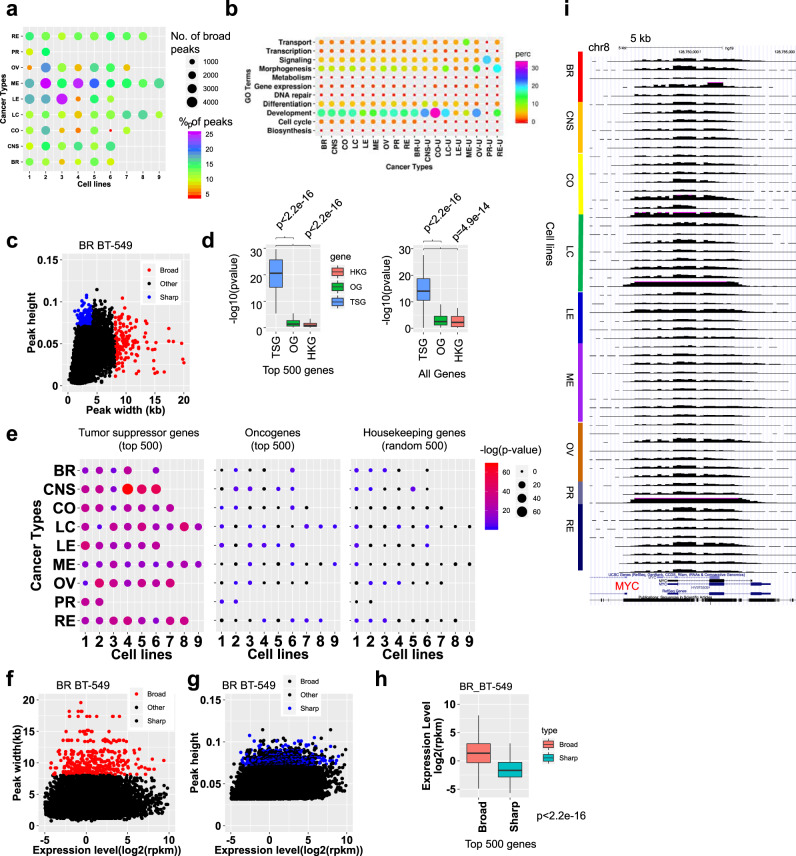
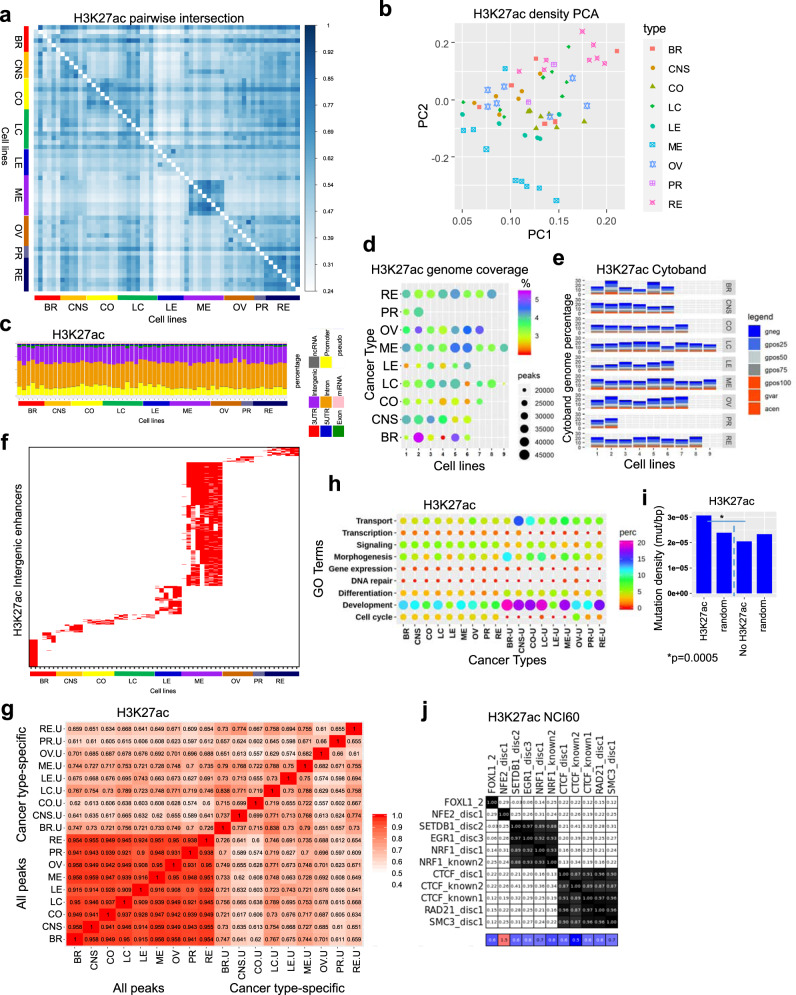
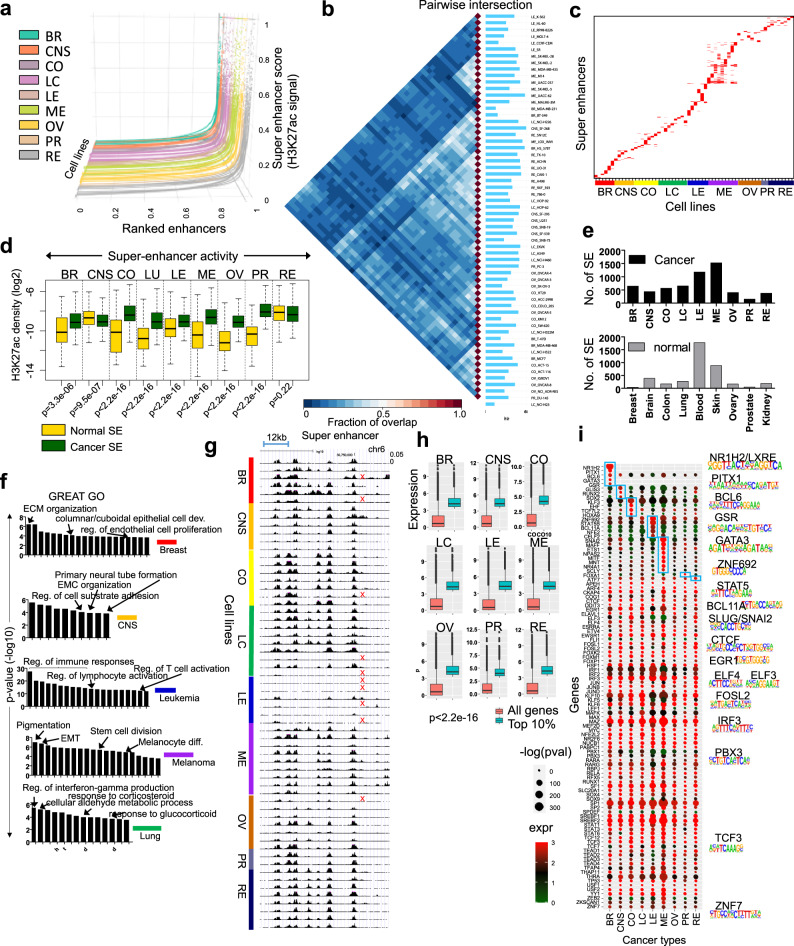
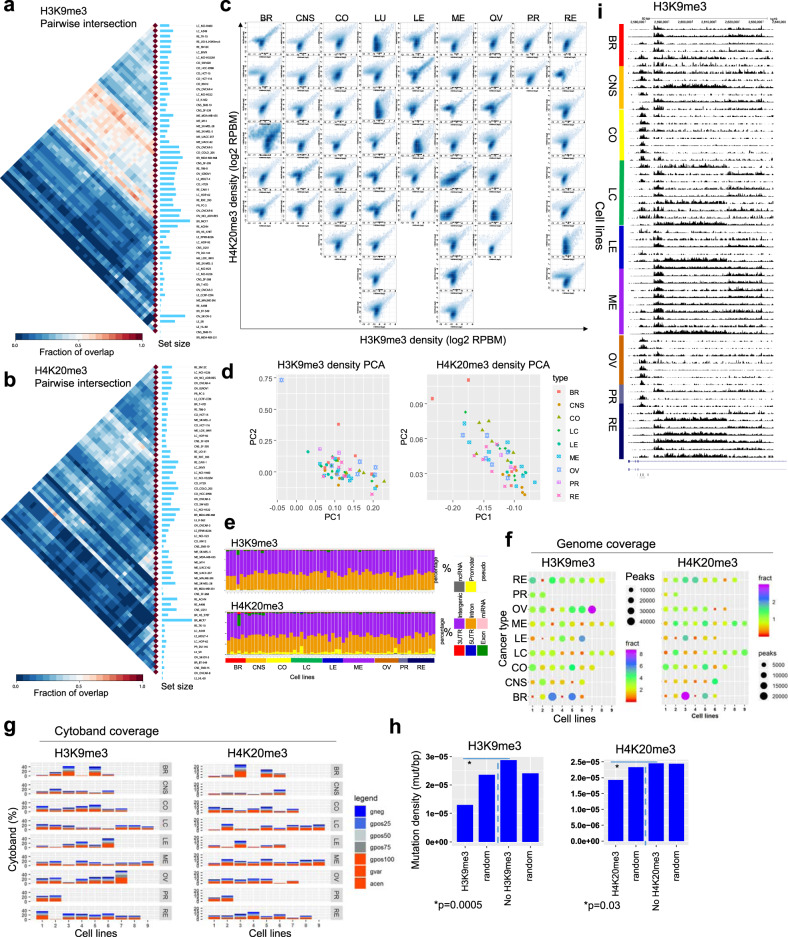
Epigenetic mechanisms contribute to the initiation and development of cancer, and epigenetic variation promotes dynamic gene expression patterns that facilitate tumor evolution and adaptation. While the NCI-60 panel represents a diverse set of human cancer cell lines that has been used to screen chemical compounds, a comprehensive epigenomic atlas of these cells has been lacking. Here, we report an integrative analysis of 60 human cancer epigenomes, representing a catalog of activating and repressive histone modifications. We identify genome-wide maps of canonical sharp and broad H3K4me3 domains at promoter regions of tumor suppressors, H3K27ac-marked conventional enhancers and super enhancers, and widespread inter-cancer and intra-cancer specific variability in H3K9me3 and H4K20me3-marked heterochromatin domains. Furthermore, we identify features of chromatin states, including chromatin state switching along chromosomes, correlation of histone modification density with genetic mutations, DNA methylation, enrichment of DNA binding motifs in regulatory regions, and gene activity and inactivity. These findings underscore the importance of integrating epigenomic maps with gene expression and genetic variation data to understand the molecular basis of human cancer. Our findings provide a resource for mining epigenomic maps of human cancer cells and for identifying epigenetic therapeutic targets.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
