

A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry
- PMID: 33659415
- PMCID: PMC7842335
- DOI: 10.21769/BioProtoc.3756
A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry
Abstract
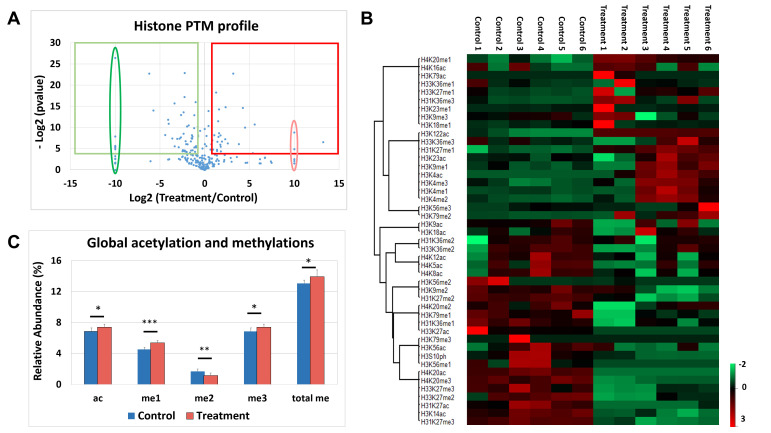
Chromatin modifications, like histone post translational modifications (PTMs), are critical for tuning gene expression and many other aspects of cell phenotype. Liquid chromatography coupled to mass spectrometry (LC-MS) has become the most suitable method to analyze histones and histone PTMs in a large-scale manner. Selected histone PTMs have known functions, and their aberrant regulation is linked to a wide variety of diseases, including cancer. However, histone analysis is scarcely used in diagnostics, partially due to the limited throughput and not ideal reproducibility of LC-MS based analysis. We describe a workflow that allows for high-throughput sample preparation is less than a day using 96-well plates. Following preparation, samples are sprayed into MS without LC, using an automated direct injection (DI-MS) method. Each analysis provides accurate quantification for 29 peptide sequences with 45 PTMs (methylations, acetylations and phosphorylations) for a total of 151 histone marks plus 16 unmodified histone peptides for relative quantification of histone variants. This workflow allows for < 1 min MS runs and higher reproducibility and robustness due to the absence of carryover or LC-based batch effects. Finally, we describe an engineered peptide sequence used to accurately monitor the efficiency of sample preparation, which can be detected during the DI-MS run.
Keywords: Advantage over Liquid-Chromatography (LC); Chromatin; Direct injection; Histone; Mass spectrometry; Post-translational modifications (PTMs).
Copyright © 2020 The Authors; exclusive licensee Bio-protocol LLC.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures






References
-
- Bannister A. J., Schneider R., Myers F. A., Thorne A. W., Crane-Robinson C. and Kouzarides T.(2005). Spatial distribution of di-and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem 280(18): 17732-17736. - PubMed
-
- Bonaldi T., Imhof A. and Regula J. T.(2004). A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 4(5): 1382-1396. - PubMed
-
- Egelhofer T. A., Minoda A., Klugman S., Lee K., Kolasinska-Zwierz P., Alekseyenko A. A., Cheung M. S., Day D. S., Gadel S., Gorchakov A. A., Gu T., Kharchenko P. V., Kuan S., Latorre I., Linder-Basso D., Luu Y., Ngo Q., Perry M., Rechtsteiner A., Riddle N. C., Schwartz Y. B., Shanower G. A., Vielle A., Ahringer J., Elgin S. C., Kuroda M. I., Pirrotta V., Ren B., Strome S., Park P. J., Karpen G. H., Hawkins R. D. and Lieb J. D.(2011). An assessment of histone-modification antibody quality. Nat Struct Mol Biol 18(1): 91-93. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
